Alpha Thalassemia Markt GRÖSSEN- UND MARKTANTEILSANALYSE - WACHSTUMSTRENDS UND PROGNOSEN (2024 - 2031)
Alpha Thalassemia Markt ist segmentiert nach Art der Krankheit (Transfusion-Dependent Alpha Thalassemia (TDT), Non-Transfusion Dependent Alpha Thalassemia (NTDT)), Durch ....
Alpha Thalassemia Markt Trends
Markttreiber - Erhöhung des Bewusstseins und der Forschung in genetischen Störungen wie Alpha-Thalassemia, was zu einer früheren Diagnose und verbesserten Therapieansätzen führt.
In den letzten zehn Jahren gab es eine beträchtliche Zunahme der Bewusstseinsinitiativen, die weltweit unternommen wurden, um das Bewusstsein über genetische Bluterkrankungen wie Alpha Thalassemia zu verbreiten. Mehrere gemeinnützige Organisationen und Patientenverteidigungsgruppen arbeiten intensiv daran, die allgemeine Bevölkerung sowie Gesundheitsdienstleister über die Anzeichen, Symptome und verfügbare Diagnose- und Behandlungsoptionen für solche erblichen Bedingungen zu erziehen. Darüber hinaus werden durch staatliche und private Finanzierung umfangreiche Forschungsaktivitäten von akademischen Institutionen und Pharmaunternehmen durchgeführt, um tiefere klinische Einblicke in die Pathophysiologie und die natürliche Geschichte dieser seltenen Krankheiten zu gewinnen. Dies hat das Verständnis über die Störung deutlich verbessert und bei der Früherkennung geholfen.
Routine-Screening-Programme wurden in vielen Ländern durchgeführt, um schwangere Frauen und Neugeborene für gemeinsame Hämoglobinopathien zu testen. Die Vorabkontrolle und Diagnoseoptionen stehen auch zunehmend zur Verfügung, um erwarteten Müttern bei der Risikobewertung zu helfen und sich auf die Herausforderungen der Betreuung eines betroffenen Kindes vorzubereiten. Zusammen haben diese Bemühungen dazu geführt, dass mehr Alpha-Thalassemia-Fälle während der fetalen Phase oder frühen Kindheit selbst diagnostiziert werden, als die meisten Patienten für Jahre undiagnostiziert bleiben. Die rechtzeitige Diagnose ermöglicht eine maßgeschneiderte Pflege und Intervention von Anfang an, um Symptome zu lindern und den Zustand richtig zu verwalten. Es hilft auch, Komplikationen und gesundheitliche Auswirkungen auf lange Sicht zu reduzieren. Alle diese Faktoren im Zusammenhang mit der wachsenden Forschung und neuen Erkenntnissen tragen zu höheren Diagnoseraten und optimierten Pflegewegen für Alpha-Thalassemia-Patienten bei.
Markttreiber - Promising Pipeline Therapien zum Antrieb des Marktes
Der Alpha-Thalassemia-Behandlungsmarkt dürfte in den kommenden Jahren lukrative Wachstumschancen sehen, die durch eine Quellungspipeline neuer Drogenkandidaten, die derzeit in der Entwicklung stehen, verursacht werden. Mehrere pharmazeutische Unternehmen und Biotech-Start-ups haben dies als ein Bereich des ungenutzten Bedarfs erkannt und arbeiten aktiv an potenziellen krankheitsmodifizierenden Therapien durch verschiedene Handlungsmechanismen. Diese innovativen Behandlungsoptionen haben, wenn sie erfolgreich in zugelassene Medikamente übersetzt werden, die Möglichkeit, die bestehenden Pflegeparadigmen, insbesondere für nicht-transfusionsabhängige Fälle von Alpha Thalassemia, zu transformieren.
Einige der am weitesten fortgeschrittenen Pipeline-Therapien, die erwartet werden, um signifikante Einnahmen zu fahren, gehören Mitapivat von Agios, Etavopivat von Protagonist Therapeutics, und Luspatercept von Acceleron/Celgene. Mitapivat ist ein erstklassiger oraler kleiner Molekülpyruvat Kinase Aktivator, der vielversprechende Ergebnisse in der effektiven Behandlung der zugrunde liegenden roten Blutkörperchen Dysfunktion in Phase II-Studien gezeigt hat und derzeit in Phase III-Studie ist. Ebenso hat Etavopivat, ein weiterer oraler Pyruvatkinaseaktivator, in frühen Versuchen eine Reduktion der extravaskulären Hämolyse nachgewiesen. Luspatercept, ein Spätstadium erythroid Reifung Agent, hat Phase III erfolgreich abgeschlossen und könnte die erste FDA zugelassene Therapie speziell für nicht-transfusionsabhängige Thalassemia Patienten.
Diese neuartigen Mittel können den Patienten eine orale Behandlungsmöglichkeit bieten, um Transfusionen deutlich zu reduzieren und gesündere rote Blutkörperchen ohne regelmäßige Krankenhausbesuche zur Transfusionsunterstützung aufzubauen. Sie werden auch dazu beitragen, einen unmissverständlichen Bedarf an der Behandlung der nicht-transfusionsuntermenge von Patienten anzugehen, die bisher minimal zugelassene therapeutische Möglichkeiten zur Verfügung gestellt hatten. Dieser Pipelinefortschritt soll in den nächsten fünf Jahren und darüber hinaus ein neues Zeitalter im Alpha-Thalassemia-Management absolvieren.
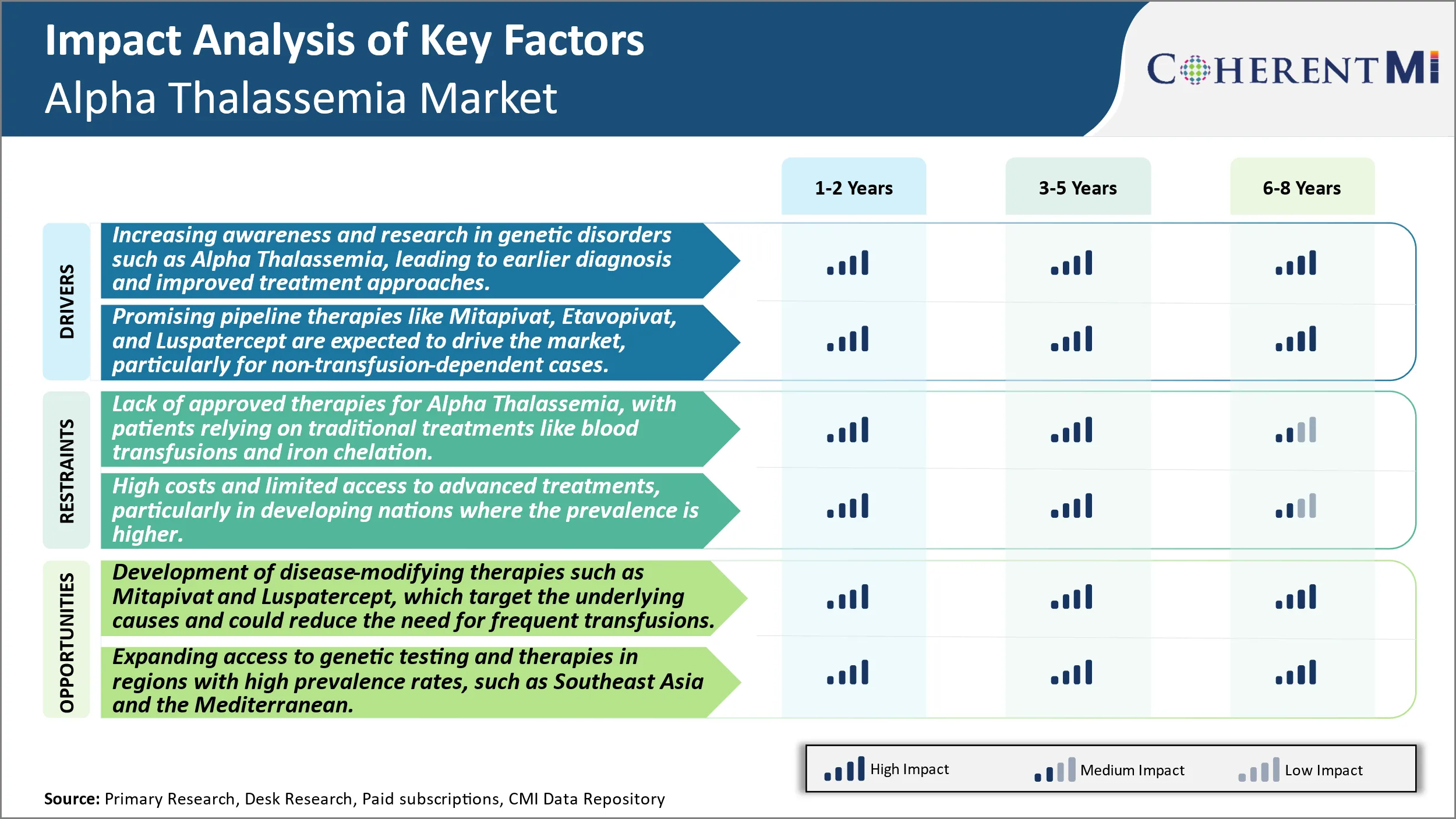
Markt-Herausforderung - Mangel an zugelassenen Therapien für Alpha-Thalassemia, mit Patienten, die auf traditionellen Behandlungen wie Bluttransfusionen und Eisen Chelation reagieren.
Der Mangel an zugelassenen krankheitsmodifizierenden Therapien für Alpha-Talassämie stellt sowohl für Patienten als auch für die Industrie eine bedeutende Herausforderung dar. Patienten sind derzeit auf unterstützende Maßnahmen wie regelmäßige Bluttransfusionen und Eisenchelat angewiesen, um schwere Anämiesymptome zu verwalten. Diese traditionellen Behandlungen sind jedoch keine Heilung und erfordern eine lebenslange Haftung, die die Lebensqualität beeinträchtigen kann. Bluttransfusionen tragen Risiken der Eisenüberlastung auf lange Sicht, wenn nicht richtig mit der chelation verwaltet. Die Bedürfnisse der regelmäßigen Überwachung des Eisenspiegels und der Verabreichung der Chelation Intravenös oder oral legt eine Behandlungsbelastung für Patienten. Der Mangel an kurativen oder krankheitsmodifizierenden Behandlungsoptionen beschränkt auf das Wachstumspotenzial. Pharmazeutische Unternehmen haben weniger Anreize, stark in die Arzneimittelforschung und -entwicklung für eine seltene Krankheit mit nur unterstützenden Pflegemöglichkeiten zu investieren. Insgesamt treibt der ungeeignete medizinische Bedarf einer Heilung sowohl Patienten- als auch Brancheninteresse an neuartigen Therapien, die die zugrunde liegende Pathologie anstreben können.
Marktchance: Entwicklung von krankheitsverändernden Therapien, um neue Möglichkeiten zu schaffen.
Es gibt vielversprechende Gelegenheit in der Entwicklung von krankheitsmodifizierenden Therapien für Alpha-Talassämie, die die Ursache für unwirksame Hämoglobinproduktion anstreben. Zwei Medikamente, Mitapivat und Luspatercept, sind in Spätstadien klinischen Studien, die ihr Potenzial zur Verringerung der Transfusionsabhängigkeit untersuchen. Mitapivat aktiviert Pyruvatkinase, die die Gesundheit und Funktion der roten Blutkörperchen wiederherstellen könnte. In klinischen Studien hat es bisher für bestimmte Patienten eine Reduzierung der Transfusionsbelastung gezeigt. Luspatercept arbeitet durch Bindung und Hemmung von Proteinen, die für unwirksame Erythropoiesis verantwortlich sind, wodurch eine normale rote Blutzellreifung ermöglicht wird. Die Genehmigung für die Beta-Talassämie hat die Grundlage für Studien in der Alpha-Talassämie gelegt. Wenn erfolgreich, können diese Therapien helfen, die klinischen Ergebnisse zu verbessern, indem weniger Transfusionen und reduzieren Eisen Überlastmanagement auf lange Sicht. Ihre Zulassung könnte den Markt neu beleben, indem sie den kritischen Bedarf an kurativen oder krankheitsmodifizierenden Behandlungen für Alpha-Talassämie-Patienten erfüllt.