Thalassemia alfa Mercato ANALISI DIMENSIONE E QUOTA - TENDENZE DI CRESCITA E PREVISIONI (2024 - 2031)
Thalassemia alfa Il mercato è segmentato da tipo di malattia (Trasfusione-Dependent Alpha Thalassemia (TDT), Non Trasfusione Dipendente Alpha Thalasse....
Thalassemia alfa Mercato Dimensione
Dimensione del mercato in USD Bn
CAGR9.10%
| Periodo di studio | 2024 - 2031 |
| CAGR | 9.10% |
| Concentrazione del mercato | Medium |
| Principali attori | Agios Farmaceutici, Novo Nordisk, Bristol-Myers Squibb, Forma terapeutica, silenzio terapeutico e tra gli altri |
faccelo sapere!
Thalassemia alfa Mercato Analisi
Il mercato globale della Thalassemia Alpha è stimato in USD 4.4 bn in 2024 e si prevede di raggiungere USD 9,1 bn entro il 2031, crescendo ad un tasso di crescita annuale composto (CAGR) del 9,10% dal 2024 al 2031. Alpha Thalassemia è un disturbo del sangue causato da mutazioni nei geni HBA1 e HBA2. La crescente prevalenza di disturbi dell'alfa-talassemia a livello globale dovrebbe guidare la crescita del mercato durante il periodo di previsione.
Il mercato sta assistendo alla crescita positiva a causa della crescente consapevolezza circa il disturbo e le sue opzioni di trattamento. Inoltre, nuovi sviluppi del prodotto per trattare le anomalie genetiche sottostanti e i programmi di screening migliorati aiutano a rilevare i casi stanno contribuendo all'espansione del mercato. Tuttavia, l'assenza di opzioni di trattamento sintomatico e la mancanza di farmaci che modificano la malattia sono alcune delle sfide che frenano la crescita del mercato.
Thalassemia alfa Mercato Tendenze
Driver di mercato - Aumentare la consapevolezza e la ricerca nei disturbi genetici come l'alfa Thalassemia, portando a diagnosi precedenti e metodi di trattamento migliorati.
Negli ultimi dieci anni si è registrato un notevole aumento delle iniziative di sensibilizzazione intraprese a livello globale per diffondere la consapevolezza sui disturbi del sangue genetico come l'Alfa Thalassemia. Numerose organizzazioni no-profit e gruppi di advocacy pazienti hanno lavorato diligentemente per educare la popolazione generale così come fornitori di assistenza sanitaria sui segni, sintomi, e opzioni di diagnosi e trattamento disponibili per tali condizioni ereditate. Inoltre, supportato da fondi pubblici e privati, sono in corso ampie attività di ricerca svolte da istituzioni accademiche e aziende farmaceutiche per acquisire approfondimenti clinici sulla patofisiologia e la storia naturale di queste malattie rare. Questo ha significativamente migliorato la comprensione del disturbo e ha aiutato nel rilevamento precoce.
I programmi di screening di routine sono stati implementati in molti paesi per testare donne incinte e neonati per emoglobinopatie comuni. Le opzioni di screening e diagnosi prenatali sono sempre più disponibili per aiutare le madri in attesa a valutare il rischio e a prepararsi alle sfide di cura di un bambino colpito. Insieme, questi sforzi hanno portato a più casi di Thalassemia Alpha diagnosticati durante la fase fetale o la prima infanzia stessa rispetto al passato quando la maggior parte dei pazienti sarebbe rimasto non diagnosticato per anni. La diagnosi tempestiva consente cure e interventi su misura dall'inizio per alleviare i sintomi e gestire correttamente la condizione. Aiuta anche a ridurre le complicazioni e gli impatti sulla salute a lungo termine. Tutti questi fattori relativi alla crescita della ricerca e nuove intuizioni sono positivamente contribuendo a tassi di diagnosi più elevati e percorsi di cura ottimizzati per i pazienti Alpha Thalassemia.
Driver di mercato - Promising Pipeline Terapie per guidare il mercato
Il mercato del trattamento di Alpha Thalassemia è probabile che le opportunità di crescita lucrativa nei prossimi anni spinte da una pipeline di gonfiore di nuovi candidati droga attualmente in fase di sviluppo. Molteplici aziende farmaceutiche e start-up biotecnologiche lo hanno riconosciuto come un'area di necessità insoddisfacente e stanno attivamente lavorando su potenziali terapie che modificano le malattie attraverso diversi meccanismi di azione. Se tradotto con successo in farmaci approvati, queste innovative opzioni di trattamento hanno la capacità di trasformare i paradigmi di cura esistenti, in particolare per casi non transfusion-dipendenti di Alpha Thalassemia.
Alcune delle terapie conduttive più avanzate che dovrebbero guidare ricavi significativi includono Mitapivat di Agios, Etavopivat di Protagonist Therapeutics, e Luspatercept di Acceleron/Celgene. Mitapivat è un attivatore di chinasi piruvato di piccola molecola orale di prima classe che ha dimostrato risultati promettenti nel trattamento efficace della disfunzione del globulo rosso sottostante negli studi di Fase II ed è attualmente in fase III di prova. Allo stesso modo, Etavopivat, un altro attivatore di chinasi piruvato orale, ha dimostrato la riduzione dell'emolisi extravascolare nelle prove iniziali. Luspatercept, un agente di maturazione dell'eritroide di fine fase, ha completato il successo della fase III e potrebbe essere la prima terapia approvata dalla FDA specificamente destinata ai pazienti affetti da talasemia non trasfusione.
Se approvato, questi nuovi agenti possono offrire ai pazienti un'opzione di trattamento orale per ridurre significativamente le trasfusioni e costruire globuli rossi più sani senza visite ospedaliere regolari per il supporto alla trasfusione. Aiuteranno inoltre ad affrontare una necessità insoddisfacente per il trattamento del sottoinsieme non trasfusione di pazienti che fino ad ora avevano opzioni terapeutiche approvate minime disponibili. Si prevede che i progressi della pipeline prevedano una nuova età nella gestione di Alpha Thalassemia nei prossimi cinque anni e oltre.
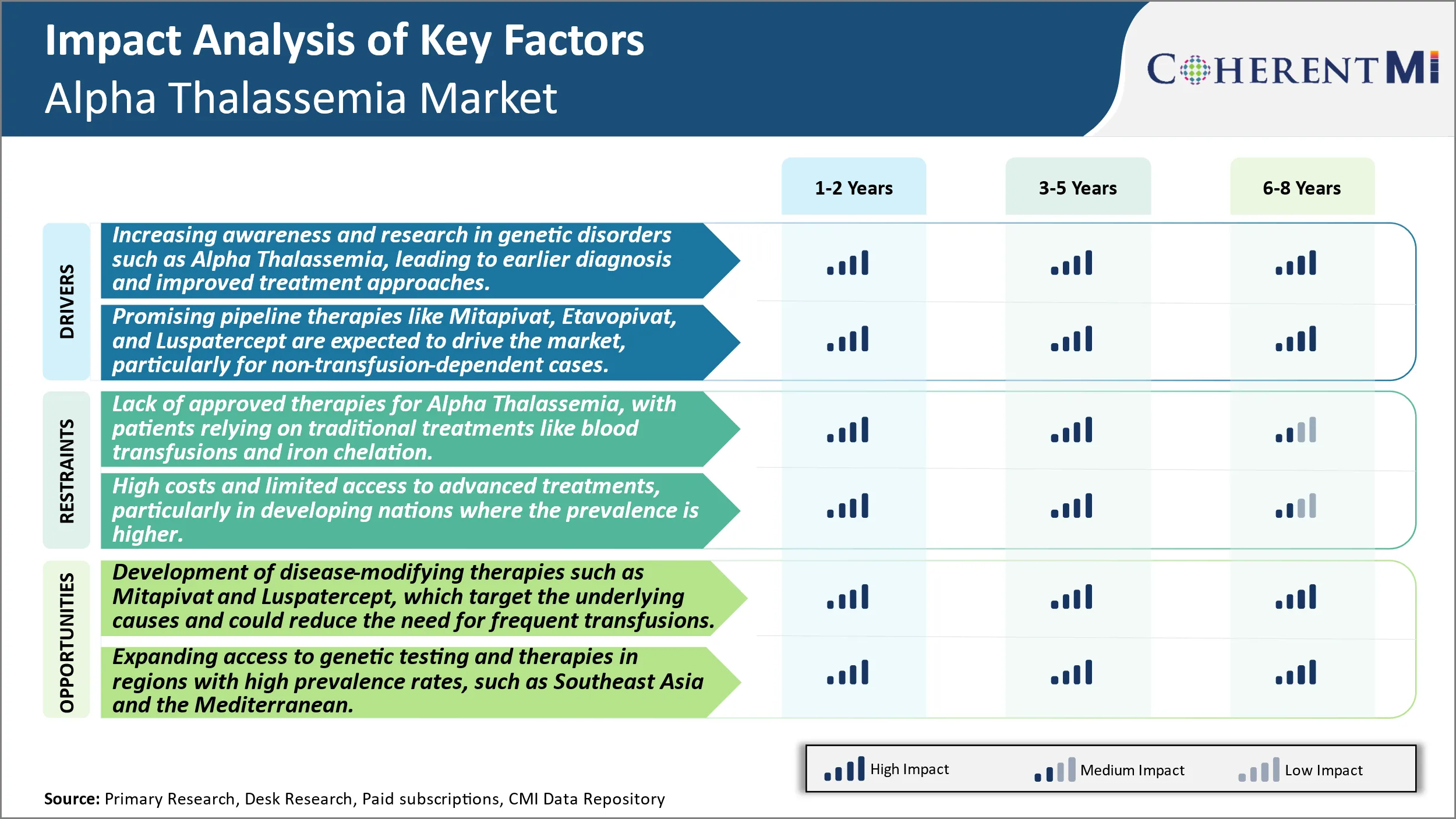
Market Challenge - Mancanza di terapie approvate per Alpha Thalassemia, con i pazienti che si basano sui trattamenti tradizionali come le trasfusioni di sangue e la chelazione di ferro.
La mancanza di terapie anti-malattie approvate per l'alfa thalassemia pone una sfida significativa sia per i pazienti che per l'industria. I pazienti sono attualmente dipendenti da misure di supporto come trasfusioni di sangue regolari e chelazione di ferro per gestire i sintomi di anemia grave. Tuttavia, questi trattamenti tradizionali non sono una cura e richiedono l'adesione permanente che può influenzare la qualità della vita. Trasfusioni di sangue portano rischi di sovraccarico di ferro a lungo termine se non gestito correttamente con chelazione. Le esigenze di monitoraggio regolare dei livelli di ferro e di somministrazione della chelazione Per via endovenosa o oralmente pone un carico di trattamento sui pazienti. Da un punto di vista industriale, la mancanza di opzioni di trattamento curative o modificanti dalle malattie limita il potenziale di crescita. Le aziende farmaceutiche hanno meno incentivi per investire pesantemente nella ricerca e nello sviluppo di farmaci per una malattia rara con solo opzioni di assistenza a disposizione. Nel complesso, la necessità medica non misurata di una cura spinge sia l'interesse del paziente che dell'industria a nuove terapie che possono colpire la patologia sottostante.
Opportunità di mercato: sviluppo di terapie per la modifica delle malattie per creare nuove opportunità.
Vi è una promettente opportunità nello sviluppo di terapie che modificano le malattie per la talassemia alfa che mira alla causa principale della produzione emoglobina inefficace. Due farmaci, Mitapivat e Luspatercept, sono in studi clinici di fine fase che indagano il loro potenziale per ridurre la dipendenza da trasfusione. Mitapivat attiva la chinasi piruvato che potrebbe ripristinare la salute e la funzione del globulo rosso. Negli studi clinici finora, ha dimostrato riduzioni nel carico di trasfusione per alcuni pazienti. Luspatercept funziona legando e inibendo le proteine responsabili dell'eritropoiesi inefficace, consentendo così la normale maturazione delle cellule rosse. La sua approvazione per la beta-thalassemia ha posto le basi per gli studi in alfa thalassemia. In caso di successo, queste terapie possono contribuire a migliorare i risultati clinici consentendo meno trasfusioni e riducendo la gestione del sovraccarico di ferro nel lungo termine. La loro approvazione potrebbe rinvigorire il mercato soddisfacendo la necessità critica di trattamenti curativi o mutevoli per pazienti alfa thalassemia.
Preferenze dei prescrittori di Thalassemia alfa Mercato
Alpha thalassemia è generalmente trattato lungo due linee principali di trattamento - prima linea e seconda linea. Nel trattamento di prima linea, la maggior parte dei prescritti preferiscono l'idrossiurea per casi da lievi a moderati. Hydroxyurea, commercializzato sotto i nomi di marca Hydrea e Droxia, lavora per ridurre la proliferazione emoglobina anormale e gestire i sintomi di anemia.
Per casi più gravi che richiedono trasfusioni di sangue regolari, i prescritti scelgono comunemente la terapia chelatatrice oltre alle trasfusioni. Deferasirox (Exjade) e deferiprone (Ferriprox) sono le opzioni di chelatore di ferro preferite prescritte per prevenire l'emocromatosi causata dall'assorbimento di ferro in eccesso da trasfusioni regolari.
Se un paziente non risponde adeguatamente o diventa dipendente dalla trasfusione, i prescritti considerano i trattamenti di seconda linea. Il trapianto di midollo di ossa è consigliato ai pazienti idonei per trattare definitivamente la malattia. Tuttavia, a causa dei rischi coinvolti, i prescritti valutano accuratamente l'idoneità al trapianto in base all'età, allo stadio della malattia e alla disponibilità di un donatore corrispondente.
Per i pazienti non idonei al trapianto, i prescriventi prescrivono Luspatercept-aamt (Reblozyl), un nuovo agente di maturazione eritroide approvato, in combinazione con la migliore assistenza di supporto trasfusioni e chelazione di ferro. Reblozyl può ridurre la dipendenza da trasfusione in alcuni pazienti.
Nel complesso i prescritti cercano trattamenti che alleviano i sintomi dell'anemia, riducono i requisiti di trasfusione, e considerano i singoli fattori del paziente come gravità, tasso di progressione e risposta alle terapie precedenti. Questo aiuta a determinare la linea più adatta e il regime di trattamento per ogni caso alfa thalassemia.
Analisi delle opzioni di trattamento di Thalassemia alfa Mercato
La talassemia alfa ha diversi livelli di gravità a seconda del numero di geni della globina alfa difettosi. I piani di trattamento sono personalizzati in base al tipo specifico. In forme lievi, nessun trattamento può essere necessario in quanto i pazienti sono spesso asintomatici. In forme moderate, la prima linea di trattamento è l'integrazione acido folico, che aiuta a promuovere la produzione di globuli rossi. Per forme gravi, le trasfusioni di sangue regolari sono tenuti a gestire i sintomi di anemia. Il trattamento preferito è trasfusioni di globuli rossi ogni 2-5 settimane a seconda dell'individuo. Questo aiuta a ridurre altre complicazioni mantenendo livelli emoglobina coerenti. La terapia di chelazione con deferoxamine è anche dato per rimuovere il ferro in eccesso dalle trasfusioni precedenti. Per il tipo più grave chiamato malattia di HbH, il trapianto di midollo osseo è l'unica opzione curativa. Tuttavia, si tratta di rischi e una perfetta partita di donatori è necessaria. Il trapianto sostituisce il midollo osseo difettoso con cellule di donatore sane. Farmaci come busulfan e ciclofosfamide sono forniti come condizionamento pre-trapianto per eliminare le cellule difettose.
Per i pazienti che non possono subire trapianti, la gestione si concentra sulle trasfusioni di sangue per tutta la vita insieme alla terapia di chelazione per evitare gravi danni agli organi da sovraccarico di ferro. Il monitoraggio ravvicinato aiuta a determinare i programmi di trasfusione ottimali su misura per ogni paziente. Nel complesso, è necessario un approccio di cura multidisciplinare per gestire efficacemente la condizione e migliorare la qualità della vita in base al livello di gravità.
Strategie vincenti chiave adottate dai principali attori di Thalassemia alfa Mercato
Adozione di Gene Terapia: Bluebird Bio ha adottato una strategia di terapia genica per Alpha Thalassemia nel 2019. Hanno sviluppato LentiGlobin, un trattamento di terapia genica che mira al difetto genetico sottostante che causa l'alfa Thalassemia. Negli studi clinici, LentiGlobin ha dimostrato una significativa indipendenza trasfusione nei pazienti, eliminando o riducendo la loro dipendenza dalle trasfusioni di sangue regolari. Questo innovativo approccio di terapia genica ha trasformato il paesaggio di trattamento per Alpha Thalassemia. Ha ricevuto l'approvazione di marketing in Europa nel 2019, rendendola la prima terapia genica approvata per la malattia.
Strategia di acquisizione: Celgene ha perseguito una strategia di acquisizione, acquisendo Agios Pharmaceuticals nel 2018 per USD7 bn. Agios aveva un farmaco sperimentale chiamato mitapivat nelle prove di fase 3 per la Thalassemia. Ciò ha permesso a Celgene di accedere a un promettente asset pipeline di fine stadio per Alpha Thalassemia. Nel 2020, mitapivat è stato approvato dalla FDA per la Thalassemia, diventando la prima malattia approvata che modifica la terapia per ridurre l'onere della trasfusione per le popolazioni non beta thalassemia pazienti.
Collaborazioni e partenariati: Global Blood Therapeutics ha collaborato con farmacie speciali e no-profit focalizzati sulle malattie rare per espandere l'accesso al loro prodotto approvato dalla FDA Oxbryta per SCD. Hanno fornito aiuti finanziari e programmi di assistenza co-pay per i pazienti idonei di Alpha Thalassemia. Ciò ha aiutato a superare le barriere di convenienza e l'assorbimento di spinta di Oxbryta, che riduce l'anemia e la dipendenza dalle trasfusioni di sangue. Prescrizioni per Oxbryta nell'indicazione Alpha Thalassemia raddoppiata nel 2021 rispetto al 2020.
Questi esempi dimostrano strategie di successo come l'innovazione della terapia genica, acquisizioni mirate e partnership per espandere l'accesso e il rimborso.
Analisi segmentale di Thalassemia alfa Mercato
Insights, per tipo di malattia: il bisogno crescente di trasfusione-dipendente Alpha Thalassemia (TDT) è evidente.
Per tipo di malattia, la Thalassemia Alfa Transfusion-Dependent (TDT) dovrebbe contribuire al 52,5% nel 2024 a causa della natura critica della cura richiesta dai pazienti. TDT è una forma grave che richiede trasfusioni di sangue regolari per tutta la vita per la sopravvivenza. I pazienti sperimentano l'anemia grave e altre complicazioni se le trasfusioni non sono adeguatamente gestite. Questo rende il trattamento non negoziabile e una priorità sanitaria primaria. Le famiglie dei pazienti TDT affrontano enormi oneri medici e finanziari per sostenere terapie di trasfusione a lungo termine. Le sfide psicosociali sono anche prevalenti a causa della dipendenza dai servizi medici. Di conseguenza, i sistemi sanitari sono invitati a ottimizzare l'accesso al sangue di qualità e investire in programmi di trasfusione per prevenire la mortalità.

Insights, per tipo di trattamento, Iron Chelation Therapy è pronto a registrare una crescita notevole negli anni a venire.
Per tipo di trattamento, Chelazione di ferro La terapia dovrebbe contribuire al 36,1% nel 2024 a causa della sua diffusa adozione come intervento frontale. Le trasfusioni affrontano efficacemente l'anemia ma introducono il sovraccarico di ferro come effetto collaterale inevitabile se lasciato non gestito. La terapia di chelazione mira a legare e rimuovere il ferro in eccesso accumulato negli organi vitali attraverso molteplici programmi di dose giornaliera. Desferrioxamine tramite infusione sottocutanea rimane un trattamento standard a livello globale nonostante la sua inconveniente. Nuovi agenti orali come deferiprone e deferasirox hanno guadagnato la preferenza a causa di una migliore conformità da somministrazione orale. Poiché le aspettative di vita dei pazienti TDT aumentano con la cura della qualità, il controllo del ferro a lungo termine ha importanza per evitare complicazioni e massimizzare il benessere.
Insights, Da parte dell'utente finale, Pediatric è previsto per Testimonianza una crescita notevole nel periodo di previsione.
Da parte dell'utente finale, Pediatric contribuisce alla quota più alta del mercato a causa di TDT spesso diagnosticato alla nascita o all'infanzia. I sintomi si manifestano dall'infanzia, richiedendo un intervento rapido. L'uso della sanità è intensivo dalle valutazioni di screening e diagnostica alle terapie di trasfusione longitudinale durante lo sviluppo. Oltre ai requisiti medici, i pazienti pediatrici hanno anche bisogno di una vasta formazione, consulenza e supporto multidisciplinare coordinato per garantire l'adesione. Gestione tempestiva tra le fasi di crescita mira a ottimizzare le pietre miliari fisiche, cognitive e sociali. Mentre la cura degli adulti rimane essenziale, la domanda pediatrica ha la precedenza per affrontare la malattia sin dall'inizio e minimizzare le conseguenze a lungo termine.
Ulteriori approfondimenti di Thalassemia alfa Mercato
Alpha Thalassemia è un disturbo genetico che colpisce la produzione di emoglobina, portando a gravi anemia e altre complicazioni. Mentre la malattia è più diffusa nei paesi in via di sviluppo, gli Stati Uniti e l'Europa stanno vedendo casi crescenti a causa della diversità della popolazione. Il mercato per la Thalassemia Alpha è in gran parte guidato da trattamenti tradizionali come trasfusioni di sangue e terapie di chelazione di ferro, ma l'emergere di nuove terapie come Mitapivat e Etavopivat offre speranza per i pazienti non trasfusioni-dipendenti. Queste nuove terapie mirano ad affrontare le cause genetiche sottostanti e migliorare la salute dei globuli rossi, che potrebbero ridurre significativamente la necessità di trasfusioni frequenti e migliorare la qualità della vita. Nonostante l'elevato potenziale di questi trattamenti, rimangono sfide, tra cui alti costi, mancanza di consapevolezza e accesso limitato alle terapie avanzate nelle regioni in via di sviluppo. Mentre la ricerca progredisce e più terapie raggiungono il mercato, il paesaggio di Alpha Thalassemia si prevede di evolvere, con migliori risultati per i pazienti e una crescita significativa nel mercato.
Panoramica competitiva di Thalassemia alfa Mercato
I principali attori che operano nel mercato di Thalassemia Alpha includono Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc, Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc.
Thalassemia alfa Mercato Leader
- Agios Farmaceutici
- Novo Nordisk
- Bristol-Myers Squibb
- Forma terapeutica
- silenzio terapeutico
Thalassemia alfa Mercato - Rivalità competitiva

Thalassemia alfa Mercato
(Dominato dai principali attori)
(Altamente competitivo con molti attori.)
Sviluppi recenti in Thalassemia alfa Mercato
- Nel gennaio 2024, Agios Pharmaceuticals annunciò risultati positivi di prova di Fase III per Mitapivat nei pazienti non dipendenti dalla trasfusione di Alpha Thalassemia, mostrando un significativo aumento dei livelli di emoglobina. L'approvazione regolamentare è prevista nel 2025.
- Nel settembre del 2022 Novo Nordisk acquisì Forma Therapeutics e ora sta sviluppando Etavopivat, un farmaco orale investigativo che mira a ridurre la necessità di trasfusioni migliorando la salute dei globuli rossi nei pazienti di Alpha Thalassemia.
Thalassemia alfa Mercato Segmentazione
- Per tipo di malattia
- Thalassemia alfa trasfusione-dipendente (TDT)
- Thalassemia Alfa Dipendente (NTDT)
- Tipo di trattamento
- Chelazione di ferro Terapia
- Trasfusioni di sangue
- Bone Marrow Trapianti
- Da parte dell'utente finale
- Pediatria
- Adulto

Vorresti esplorare l'opzione di acquistosingole sezioni di questo report?
Domande frequenti :
Quanto è grande il mercato dell'Alfa Thalassemia?
Il mercato Global Alpha Thalassemia è stimato essere valutato a USD 4.4 bn nel 2024 e si prevede di raggiungere USD 9,1 bn entro il 2031.
Quale sarà il CAGR del Mercato Alfa Thalassemia?
Il CAGR dell'Alfa Thalassemia Il mercato è previsto per il 9,10% dal 2024-2031.
Quali sono i fattori chiave che ostacolano la crescita dell'Alfa Thalassemia Market?
La mancanza di terapie approvate per l'alfa thalassemia, con i pazienti che si affidano a trattamenti tradizionali come trasfusioni di sangue e chelazione di ferro e alti costi e accesso limitato a trattamenti avanzati, in particolare nelle nazioni in via di sviluppo dove la prevalenza è più alta sono i principali fattori che ostacolano la crescita del mercato dell'alfa Thalassemia.
Quali sono i principali fattori che guidano la crescita del mercato di Thalassemia Alpha?
La crescente consapevolezza e la ricerca nei disturbi genetici come l'alfa thalassemia, che porta alla diagnosi precedente e ai migliori approcci di trattamento e promettenti terapie pipeline come Mitapivat, Etavopivat, e Luspatercept sono tenuti a guidare il mercato, in particolare per i casi non transfusion-dipendenti sono i principali fattori che guidano il mercato di Thalassemia Alpha.
Qual è il principale tipo di malattia nel mercato della talasemia alfa?
La Thalassemia Alfa Dipendente non Trasfusione (NTDT) è il segmento leader del tipo di malattia.
Quali sono i principali attori che operano nel mercato della Thalassemia Alpha?
Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc, Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc, Astrazeneca, F.Hoffman La Roche, Jansen Pharma