Alpha Thalassemia Mercado ANÁLISE DE TAMANHO E PARTICIPAÇÃO - TENDÊNCIAS DE CRESCIMENTO E PREVISÕES (2024 - 2031)
Alpha Thalassemia O mercado é segmentado por tipo de doença (Transfusion-Dependent Alpha Thalassemia (TDT), não-Transfusion Dependente Alpha Thalassem....
Alpha Thalassemia Mercado Tendências
Driver de mercado - Aumentando a conscientização e a pesquisa em distúrbios genéticos, como Alpha Thalassemia, levando ao diagnóstico anterior e melhores abordagens de tratamento.
Ao longo da última década houve um aumento substancial nas iniciativas de conscientização realizadas globalmente para espalhar a conscientização sobre os transtornos genéticos do sangue como Alpha Thalassemia. Várias organizações sem fins lucrativos e grupos de defesa do paciente têm trabalhado diligentemente para educar a população geral, bem como prestadores de cuidados de saúde sobre os sinais, sintomas e opções de diagnóstico e tratamento disponíveis para tais condições herdadas. Além disso, apoiado pelo governo e financiamento privado, extensas atividades de pesquisa estão sendo realizadas por instituições acadêmicas e empresas farmacêuticas para obter insights clínicos mais profundos sobre a fisiopatologia e história natural dessas doenças raras. Isso melhorou significativamente a compreensão sobre o transtorno e ajudou na detecção precoce.
Programas de rastreamento de rotina foram implementados em muitos países para testar mulheres grávidas e recém-nascidos para hemoglobinopatias comuns. As opções de rastreamento e diagnóstico pré-natal também estão cada vez mais disponíveis para ajudar as mães grávidas a avaliar o risco e preparar os desafios de cuidar de uma criança afetada. Juntos, esses esforços levaram a mais casos de Thalassemia Alfa sendo diagnosticados durante a fase fetal ou a própria infância em comparação com o passado quando a maioria dos pacientes permaneceria não diagnosticada por anos. O diagnóstico oportuna permite cuidados e intervenções sob medida do início para aliviar os sintomas e gerenciar a condição corretamente. Também ajuda a reduzir complicações e impactos de saúde no longo prazo. Todos esses fatores relacionados ao crescimento da pesquisa e novos insights estão contribuindo positivamente para maiores taxas de diagnóstico e vias de atendimento otimizadas para pacientes da Thalassemia Alpha.
Driver de mercado - Promising Pipeline Therapies para dirigir o mercado
O mercado de tratamento Alpha Thalassemia é susceptível de ver oportunidades de crescimento lucrativo nos próximos anos impulsionado por um pipeline de inchaço de novos candidatos à droga atualmente em desenvolvimento. Várias empresas farmacêuticas e start-ups biotecnológicos reconheceram isso como uma área de necessidade não satisfeita e estão trabalhando ativamente em potenciais terapias modificadoras de doenças através de diversos mecanismos de ação. Se traduzidas com sucesso em medicamentos aprovados, essas opções de tratamento inovadoras têm a capacidade de transformar os paradigmas existentes de cuidado, particularmente para casos não-transfusion-dependentes de Alpha Thalassemia.
Algumas das terapias de pipeline mais avançadas que são esperados para gerar receitas significativas incluem Mitapivat por Agios, Etavopivat por Protagonist Therapeutics, e Luspatercept por Acceleron/Celgene. Mitapivat é um ativador pyruvate kinase pyruvate da molécula pequena oral, que mostrou resultados promissores no tratamento eficaz da disfunção da célula vermelha subjacente nos estudos da fase II e está atualmente no teste da fase III. Da mesma forma, Etavopivat, outro ativador pyruvate kinase oral, demonstrou redução na hemolise extravascular em ensaios iniciais. Luspatercept, um agente de maturação eritroide investigacional de última fase, concluiu a Fase III com sucesso e poderia ser a primeira terapia aprovada pela FDA especificamente destinada a pacientes não-transfusão-dependentes da Thalassemia.
Se aprovado, esses novos agentes podem oferecer aos pacientes uma opção de tratamento oral para reduzir significativamente as transfusões e construir células sanguíneas vermelhas mais saudáveis sem visitas hospitalares regulares para apoio à transfusão. Eles também ajudarão a abordar uma necessidade não satisfeita de tratar o subconjunto não-transfusão de pacientes que até agora tinham opções terapêuticas aprovadas mínimas disponíveis. Este progresso do pipeline é esperado para anunciar uma nova idade na gestão Alpha Thalassemia ao longo dos próximos cinco anos e além.
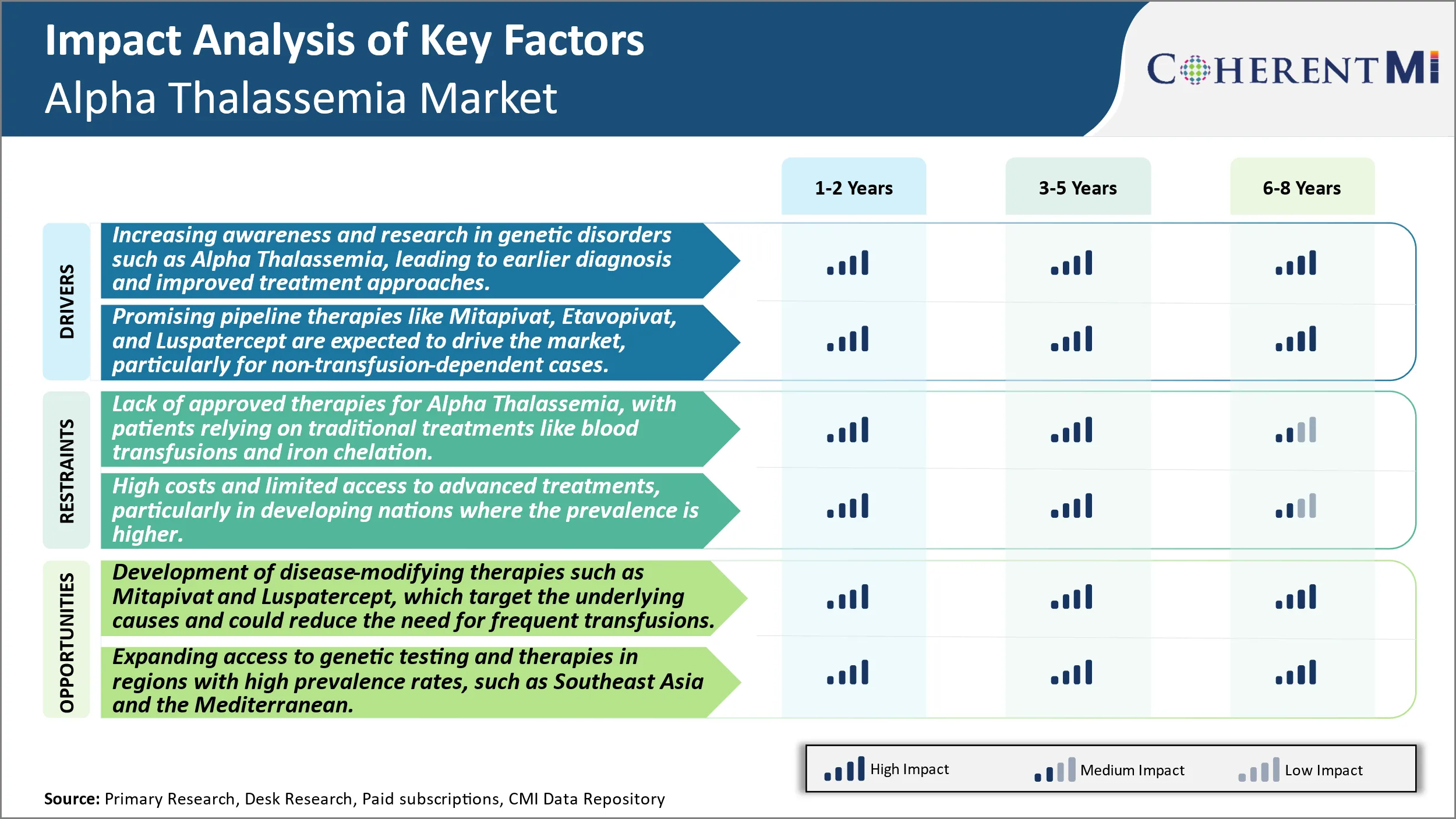
Desafio de Mercado - Falta de Terapias Aprovadas para a Thalassemia Alfa, Com Pacientes Baseando-se em Tratamentos Tradicionais Como Transfusões de Sangue e Chelation de Ferro.
A falta de terapias homologadas para a talassemia alfa representa um desafio significativo para ambos os pacientes e para a indústria. Atualmente, os pacientes dependem de medidas de apoio, como transfusões sanguíneas regulares e quilatação de ferro para gerenciar sintomas graves de anemia. No entanto, esses tratamentos tradicionais não são uma cura e exigem a adesão ao longo da vida que pode impactar a qualidade de vida. As transfusões de sangue carregam riscos de sobrecarga de ferro no longo prazo, se não forem geridas corretamente com a quilatação. As necessidades de controlo regular dos níveis de ferro e de gestão da quilatação Intravenamente ou por via oral coloca uma carga de tratamento em pacientes. De uma perspectiva da indústria, a falta de opções curativas ou de tratamento modificador de doenças limita o potencial de crescimento. As empresas farmacêuticas têm menos incentivos para investir fortemente na pesquisa e desenvolvimento de drogas para uma doença rara com apenas opções de cuidados de apoio disponíveis. Em geral, a necessidade médica não satisfeita de uma cura impulsiona o interesse do paciente e da indústria em terapias novas que podem segmentar a patologia subjacente.
Oportunidade de mercado: Desenvolvimento de terapias de transformação de doenças para criar oportunidades novárias.
Há uma oportunidade promissora no desenvolvimento de terapias modificadoras de doenças para a talassemia alfa que visam a causa raiz da produção de hemoglobina ineficaz. Duas drogas, Mitapivat e Luspatercept, estão em ensaios clínicos de última fase investigando seu potencial para reduzir a dependência de transfusão. Mitapivat ativa a pyruvate kinase que poderia restaurar a saúde e função da célula vermelha do sangue. Em estudos clínicos até agora, demonstrou reduções na carga de transfusão para certos pacientes. Luspatercept funciona através da ligação e inibição de proteínas responsáveis pela eritropoiese ineficaz, permitindo assim a maturação normal das células vermelhas do sangue. Sua aprovação para beta-talassemia estabeleceu a base para estudos na talassemia alfa. Se bem sucedido, essas terapias podem ajudar a melhorar os resultados clínicos, permitindo menos transfusões e reduzindo o gerenciamento de sobrecarga de ferro ao longo do longo prazo. Sua aprovação poderia revigorar o mercado, cumprindo a necessidade crítica de tratamentos curativos ou modificadores de doenças para pacientes alfa talassemia.