Alpha Thalassemia Markt GRÖSSEN- UND MARKTANTEILSANALYSE - WACHSTUMSTRENDS UND PROGNOSEN (2024 - 2031)
Alpha Thalassemia Markt ist segmentiert nach Art der Krankheit (Transfusion-Dependent Alpha Thalassemia (TDT), Non-Transfusion Dependent Alpha Thalass....
Alpha Thalassemia Markt Größe
Marktgröße in USD Bn
CAGR9.10%
| Studienzeitraum | 2024 - 2031 |
| CAGR | 9.10% |
| Marktkonzentration | Medium |
| Wichtige Akteure | Agios Pharmazeutika, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeuten, Stille Therapie und unter anderem |
Bitte lassen Sie es uns wissen!
Alpha Thalassemia Markt Analyse
Der Markt für Global Alpha Thalassemia wird geschätzt auf USD 4,4 Mrd. in 2024 und wird voraussichtlich erreichen USD 9,1 Mrd.Wachstumsrate (CAGR) von 9,10% von 2024 bis 2031. Alpha Thalassemia ist eine Blutkrankheit, die durch Mutationen in den HBA1- und HBA2-Genen verursacht wird. Die zunehmende Prävalenz von Alpha-Talassämie-Erkrankungen weltweit wird erwartet, dass das Wachstum des Marktes während der Prognosezeit vorangetrieben wird.
Der Markt erlebt ein positives Wachstum aufgrund des steigenden Bewusstseins über die Störung und seine Behandlungsoptionen. Darüber hinaus tragen neue Produktentwicklungen zur Behandlung der zugrunde liegenden genetischen Abnormitäten und verbesserten Screening-Programme dazu bei, Fälle zu erkennen, die zur Markterweiterung beitragen. Allerdings sind das Fehlen symptomatischer Behandlungsoptionen und das Fehlen von krankheitsmodifizierenden Medikamenten einige der Herausforderungen, die das Marktwachstum zurückhalten.
Alpha Thalassemia Markt Trends
Markttreiber - Erhöhung des Bewusstseins und der Forschung in genetischen Störungen wie Alpha-Thalassemia, was zu einer früheren Diagnose und verbesserten Therapieansätzen führt.
In den letzten zehn Jahren gab es eine beträchtliche Zunahme der Bewusstseinsinitiativen, die weltweit unternommen wurden, um das Bewusstsein über genetische Bluterkrankungen wie Alpha Thalassemia zu verbreiten. Mehrere gemeinnützige Organisationen und Patientenverteidigungsgruppen arbeiten intensiv daran, die allgemeine Bevölkerung sowie Gesundheitsdienstleister über die Anzeichen, Symptome und verfügbare Diagnose- und Behandlungsoptionen für solche erblichen Bedingungen zu erziehen. Darüber hinaus werden durch staatliche und private Finanzierung umfangreiche Forschungsaktivitäten von akademischen Institutionen und Pharmaunternehmen durchgeführt, um tiefere klinische Einblicke in die Pathophysiologie und die natürliche Geschichte dieser seltenen Krankheiten zu gewinnen. Dies hat das Verständnis über die Störung deutlich verbessert und bei der Früherkennung geholfen.
Routine-Screening-Programme wurden in vielen Ländern durchgeführt, um schwangere Frauen und Neugeborene für gemeinsame Hämoglobinopathien zu testen. Die Vorabkontrolle und Diagnoseoptionen stehen auch zunehmend zur Verfügung, um erwarteten Müttern bei der Risikobewertung zu helfen und sich auf die Herausforderungen der Betreuung eines betroffenen Kindes vorzubereiten. Zusammen haben diese Bemühungen dazu geführt, dass mehr Alpha-Thalassemia-Fälle während der fetalen Phase oder frühen Kindheit selbst diagnostiziert werden, als die meisten Patienten für Jahre undiagnostiziert bleiben. Die rechtzeitige Diagnose ermöglicht eine maßgeschneiderte Pflege und Intervention von Anfang an, um Symptome zu lindern und den Zustand richtig zu verwalten. Es hilft auch, Komplikationen und gesundheitliche Auswirkungen auf lange Sicht zu reduzieren. Alle diese Faktoren im Zusammenhang mit der wachsenden Forschung und neuen Erkenntnissen tragen zu höheren Diagnoseraten und optimierten Pflegewegen für Alpha-Thalassemia-Patienten bei.
Markttreiber - Promising Pipeline Therapien zum Antrieb des Marktes
Der Alpha-Thalassemia-Behandlungsmarkt dürfte in den kommenden Jahren lukrative Wachstumschancen sehen, die durch eine Quellungspipeline neuer Drogenkandidaten, die derzeit in der Entwicklung stehen, verursacht werden. Mehrere pharmazeutische Unternehmen und Biotech-Start-ups haben dies als ein Bereich des ungenutzten Bedarfs erkannt und arbeiten aktiv an potenziellen krankheitsmodifizierenden Therapien durch verschiedene Handlungsmechanismen. Diese innovativen Behandlungsoptionen haben, wenn sie erfolgreich in zugelassene Medikamente übersetzt werden, die Möglichkeit, die bestehenden Pflegeparadigmen, insbesondere für nicht-transfusionsabhängige Fälle von Alpha Thalassemia, zu transformieren.
Einige der am weitesten fortgeschrittenen Pipeline-Therapien, die erwartet werden, um signifikante Einnahmen zu fahren, gehören Mitapivat von Agios, Etavopivat von Protagonist Therapeutics, und Luspatercept von Acceleron/Celgene. Mitapivat ist ein erstklassiger oraler kleiner Molekülpyruvat Kinase Aktivator, der vielversprechende Ergebnisse in der effektiven Behandlung der zugrunde liegenden roten Blutkörperchen Dysfunktion in Phase II-Studien gezeigt hat und derzeit in Phase III-Studie ist. Ebenso hat Etavopivat, ein weiterer oraler Pyruvatkinaseaktivator, in frühen Versuchen eine Reduktion der extravaskulären Hämolyse nachgewiesen. Luspatercept, ein Spätstadium erythroid Reifung Agent, hat Phase III erfolgreich abgeschlossen und könnte die erste FDA zugelassene Therapie speziell für nicht-transfusionsabhängige Thalassemia Patienten.
Diese neuartigen Mittel können den Patienten eine orale Behandlungsmöglichkeit bieten, um Transfusionen deutlich zu reduzieren und gesündere rote Blutkörperchen ohne regelmäßige Krankenhausbesuche zur Transfusionsunterstützung aufzubauen. Sie werden auch dazu beitragen, einen unmissverständlichen Bedarf an der Behandlung der nicht-transfusionsuntermenge von Patienten anzugehen, die bisher minimal zugelassene therapeutische Möglichkeiten zur Verfügung gestellt hatten. Dieser Pipelinefortschritt soll in den nächsten fünf Jahren und darüber hinaus ein neues Zeitalter im Alpha-Thalassemia-Management absolvieren.
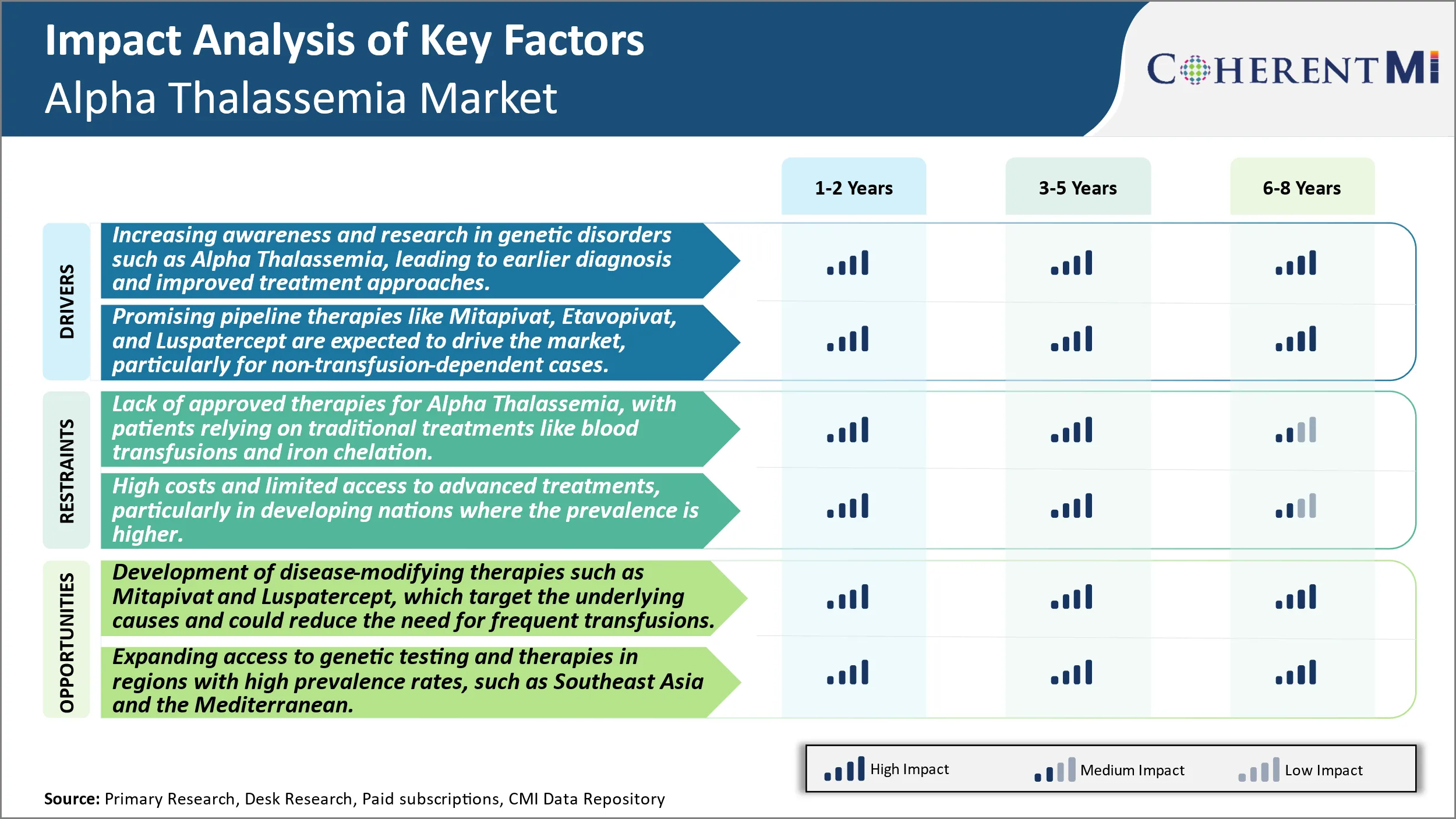
Markt-Herausforderung - Mangel an zugelassenen Therapien für Alpha-Thalassemia, mit Patienten, die auf traditionellen Behandlungen wie Bluttransfusionen und Eisen Chelation reagieren.
Der Mangel an zugelassenen krankheitsmodifizierenden Therapien für Alpha-Talassämie stellt sowohl für Patienten als auch für die Industrie eine bedeutende Herausforderung dar. Patienten sind derzeit auf unterstützende Maßnahmen wie regelmäßige Bluttransfusionen und Eisenchelat angewiesen, um schwere Anämiesymptome zu verwalten. Diese traditionellen Behandlungen sind jedoch keine Heilung und erfordern eine lebenslange Haftung, die die Lebensqualität beeinträchtigen kann. Bluttransfusionen tragen Risiken der Eisenüberlastung auf lange Sicht, wenn nicht richtig mit der chelation verwaltet. Die Bedürfnisse der regelmäßigen Überwachung des Eisenspiegels und der Verabreichung der Chelation Intravenös oder oral legt eine Behandlungsbelastung für Patienten. Der Mangel an kurativen oder krankheitsmodifizierenden Behandlungsoptionen beschränkt auf das Wachstumspotenzial. Pharmazeutische Unternehmen haben weniger Anreize, stark in die Arzneimittelforschung und -entwicklung für eine seltene Krankheit mit nur unterstützenden Pflegemöglichkeiten zu investieren. Insgesamt treibt der ungeeignete medizinische Bedarf einer Heilung sowohl Patienten- als auch Brancheninteresse an neuartigen Therapien, die die zugrunde liegende Pathologie anstreben können.
Marktchance: Entwicklung von krankheitsverändernden Therapien, um neue Möglichkeiten zu schaffen.
Es gibt vielversprechende Gelegenheit in der Entwicklung von krankheitsmodifizierenden Therapien für Alpha-Talassämie, die die Ursache für unwirksame Hämoglobinproduktion anstreben. Zwei Medikamente, Mitapivat und Luspatercept, sind in Spätstadien klinischen Studien, die ihr Potenzial zur Verringerung der Transfusionsabhängigkeit untersuchen. Mitapivat aktiviert Pyruvatkinase, die die Gesundheit und Funktion der roten Blutkörperchen wiederherstellen könnte. In klinischen Studien hat es bisher für bestimmte Patienten eine Reduzierung der Transfusionsbelastung gezeigt. Luspatercept arbeitet durch Bindung und Hemmung von Proteinen, die für unwirksame Erythropoiesis verantwortlich sind, wodurch eine normale rote Blutzellreifung ermöglicht wird. Die Genehmigung für die Beta-Talassämie hat die Grundlage für Studien in der Alpha-Talassämie gelegt. Wenn erfolgreich, können diese Therapien helfen, die klinischen Ergebnisse zu verbessern, indem weniger Transfusionen und reduzieren Eisen Überlastmanagement auf lange Sicht. Ihre Zulassung könnte den Markt neu beleben, indem sie den kritischen Bedarf an kurativen oder krankheitsmodifizierenden Behandlungen für Alpha-Talassämie-Patienten erfüllt.
Präferenzen der Verschreiber von Alpha Thalassemia Markt
Alpha-Talassämie wird in der Regel entlang zwei Hauptlinien der Behandlung behandelt - erste Linie und zweite Linie. Bei der Erstbehandlung bevorzugen die meisten Verschreibungen Hydroxyharnstoff für milde bis mäßige Fälle. Hydroxyurea, vermarktet unter Markennamen Hydrea und Droxia, arbeitet, um anormale Hämoglobinproliferation zu reduzieren und Anämie-Symptome zu verwalten.
Für schwerere Fälle, die regelmäßige Bluttransfusionen erfordern, wählen Verschreibungen häufig die Chelatatortherapie zusätzlich zu Transfusionen. Deferasirox (Exjade) und Deferipron (Ferriprox) sind bevorzugte Eisenchelator-Optionen vorgeschrieben, um Hämochromatose durch überschüssige Eisenaufnahme durch regelmäßige Transfusionen zu verhindern.
Wenn ein Patient nicht adäquat reagiert oder transfusionsunabhängig wird, betrachten Prescriber Zweitlinienbehandlungen. Knochenmarktransplantation wird für förderfähige Patienten empfohlen, die Krankheit endgültig zu behandeln. Aufgrund der damit verbundenen Risiken bewerten die Verschreibungen die Eignung der Transplantation basierend auf Alter, Krankheitsstufe und Verfügbarkeit eines angepassten Spenders.
Für Patienten, die nicht für Transplantate zugelassen sind, verschreiben die Verwalter Luspatercept-aamt (Reblozyl), ein neu zugelassenes Erythroid-Maturationsmittel, in Kombination mit bester unterstützender Pflege einschließlich Transfusionen und Eisenchelatierung. Reblozyl kann die Transfusionsabhängigkeit bei einigen Patienten reduzieren.
Insgesamt Beschreiber suchen Behandlungen, die Anämiesymptome mildern, Transfusionsanforderungen reduzieren und individuelle Patientenfaktoren wie Schwere, Progressionsrate und Reaktion auf vorherige Therapien berücksichtigen. Dies hilft, die am besten geeignete Linie und Behandlung für jeden Alpha-Talassämie Fall zu bestimmen.
Analyse der Behandlungsoptionen von Alpha Thalassemia Markt
Alpha-Talassämie hat je nach Anzahl der defekten Alpha-Globin-Gene unterschiedliche Schweregrade. Die Behandlungspläne sind auf der Grundlage des spezifischen Typs zugeschnitten. In milden Formen kann keine Behandlung benötigt werden, da Patienten oft asymptomatisch sind. In moderaten Formen ist die erste Linie der Behandlung Folsäure-Ergänzung, die hilft, die rote Blutzellenproduktion zu fördern. Für schwere Formen sind regelmäßige Bluttransfusionen erforderlich, um Anämie Symptome zu verwalten. Die bevorzugte Behandlung ist die rote Blutzelltransfusion alle 2-5 Wochen je nach Einzelperson. Dies hilft, andere Komplikationen zu reduzieren, indem konsistente Hämoglobin-Level beibehalten werden. Die Chelationstherapie mit Deferoxamin wird auch zur Entfernung von überschüssigem Eisen aus früheren Transfusionen gegeben. Für den schwersten Typ HbH-Krankheit ist die Knochenmarktransplantation die einzige heilende Option. Es kommt jedoch mit Risiken und ein perfektes Donor Match ist notwendig. Die Transplantation ersetzt defektes Knochenmark durch gesunde Spenderzellen. Medikamente wie Busulfan und Cyclophosphamid werden als Vortransplantation zur Eliminierung von fehlerhaften Zellen bereitgestellt.
Für Patienten, die nicht transplantiert werden können, konzentriert sich das Management auf lebenslange Bluttransfusionen zusammen mit der Chelattherapie, um schwere Organschäden durch Eisenüberlastung zu vermeiden. Eine enge Überwachung hilft, optimale Transfusionspläne für jeden Patienten zu ermitteln. Insgesamt wird ein multidisziplinärer Pflegeansatz benötigt, um den Zustand effektiv zu verwalten und die Lebensqualität basierend auf dem Schweregrad zu verbessern.
Wichtige Erfolgsstrategien der Hauptakteure von Alpha Thalassemia Markt
Annahme von Gene Therapie: Bluebird Bio verabschiedete 2019 eine Gentherapiestrategie für Alpha Thalassemia. Sie entwickelten LentiGlobin, eine Gentherapiebehandlung, die auf den zugrunde liegenden genetischen Defekt abzielt, der Alpha Thalassemia verursacht. In klinischen Studien zeigte LentiGlobin signifikante Transfusionsunabhängigkeit bei Patienten, die ihre Abhängigkeit von regelmäßigen Bluttransfusionen beseitigen oder verringern. Dieser innovative Gentherapieansatz hat die Behandlungslandschaft für Alpha Thalassemia transformiert. Es erhielt die Genehmigung für das Marketing in Europa im Jahr 2019, so dass es die erste genehmigte Gentherapie für die Krankheit.
Akquisitionsstrategie: Celgene verfolgte eine Akquisitionsstrategie und erwarb 2018 Agios Pharmaceuticals für USD7 Mrd. Agios hatte ein experimentelles Medikament namens Mitapivat in Phase 3 Studien für Thalassemia. Damit konnte Celgene Zugang zu einem vielversprechenden Pipeline-Asset für Alpha Thalassemia gewinnen. Im Jahr 2020 wurde die Mitapivat von der FDA für Thalassemia genehmigt und wurde die erste genehmigte Krankheitsmodifizierungstherapie, um Transfusionslast für nicht-beta-Talassämie-Patienten zu reduzieren.
Zusammenarbeit und Partnerschaften: Global Blood Therapeutics hat mit Spezial-Apotheken und Non-Profits zusammengearbeitet, die sich auf seltene Krankheiten konzentrierten, um den Zugang zu ihrem FDA zugelassenen Produkt Oxbryta für SCD zu erweitern. Sie boten finanzielle Hilfe- und Mitkostenhilfeprogramme für förderfähige Alpha-Thalassemia-Patienten an. Dies half, Erschwinglichkeitsbarrieren zu überwinden und die Aufnahme von Oxbryta zu beschleunigen, was die Anämie und die Abhängigkeit von Bluttransfusionen reduziert. Die Verschreibungen für Oxbryta in der Alpha-Thalassemia-Indikation verdoppelten sich im Jahr 2021 im Vergleich zu 2020.
Diese Beispiele zeigen erfolgreiche Strategien wie Gentherapie-Innovation, gezielte Akquisitionen und Partnerschaften, um Zugang und Erstattung zu erweitern.
Segmentanalyse von Alpha Thalassemia Markt
Insights, Nach Art der Krankheit: Die Growig Need for Transfusion-Dependent Alpha Thalassemia (TDT) ist offensichtlich.
Durch die Art der Krankheit wird erwartet, dass Transfusion-Dependent Alpha Thalassemia (TDT) im Jahr 2024 52,5% aufgrund der kritischen Art der Pflege, die von Patienten benötigt wird, beitragen wird. TDT ist eine schwere Form, die lebenslange regelmäßige Bluttransfusionen zum Überleben erfordert. Patienten erleben schwere Anämie und andere Komplikationen, wenn Transfusionen nicht richtig verwaltet werden. Dies macht die Behandlung nicht verhandelbar und eine primäre Gesundheitspriorität. Familien von TDT-Patienten stehen vor immensen medizinischen und finanziellen Belastungen, um langfristige Transfusionstherapien zu erhalten. Psychosoziale Herausforderungen sind auch aufgrund der Abhängigkeit von medizinischen Dienstleistungen weit verbreitet. Dadurch werden Gesundheitssysteme aufgefordert, den Zugang zu qualitätsgerechtem Blut zu optimieren und in Transfusionsprogramme zu investieren, um die Sterblichkeit zu verhindern.

Insights, nach Behandlungsart, ist die Eiserne Chelation Therapy Poised, um ein bemerkenswertes Wachstum in den kommenden Jahren zu registrieren.
Durch Behandlung Typ, Eisen Chelation Die Therapie soll im Jahr 2024 aufgrund ihrer weit verbreiteten Einführung als Frontintervention 36,1 % beitragen. Transfusionen adressieren effektiv Anämie, stellen aber Eisenüberlastung als unvermeidliche Nebenwirkung vor, wenn sie unbemannt bleiben. Chelation Therapie zielt darauf ab, überschüssiges Eisen in lebenswichtigen Organen durch mehrere tägliche Dosierungspläne binden und entfernen. Desferrioxamin über subkutane Infusion bleibt weltweit eine Standardbehandlung trotz seiner Unannehmlichkeiten. Neue orale Mittel wie Deferipron und Deferasirox haben aufgrund einer besseren Einhaltung der oralen Verabreichung bevorzugt. Da die Lebenserwartungen der TDT-Patienten mit der Qualitätskontrolle steigen, ist die Langzeit-Eisenkontrolle wichtig, um Komplikationen abzuwenden und das Wohlbefinden zu maximieren.
Insights, By End-User, Pediatric wird erwartet, um ein bemerkenswertes Wachstum in der Prognosezeit.
Durch End-User trägt Pediatric den höchsten Marktanteil des Marktes bei, da TDT oft bei der Geburt oder in der frühen Kindheit diagnostiziert wird. Symptome manifestieren sich durch Infancy, erfordert schnelle Intervention. Die Nutzung der Gesundheitsversorgung ist von Screening- und Diagnoseauswertungen bis hin zu Längstransfusionstherapien während der gesamten Entwicklung intensiv. Neben medizinischen Anforderungen benötigen Kinderpatienten auch umfangreiche Ausbildung, Beratung und koordinierte multidisziplinäre Unterstützung, um die Einhaltung zu gewährleisten. Zeitgemäßes Management inmitten von Wachstumsphasen zielt darauf ab, physische, kognitive und soziale Meilensteine zu optimieren. Während die Betreuung von Erwachsenen von grundlegender Bedeutung bleibt, geht die pädiatrische Nachfrage voraus, die Krankheit von Anfang an anzusprechen und langfristige Folgen zu minimieren.
Zusätzliche Einblicke von Alpha Thalassemia Markt
Alpha Thalassemia ist eine genetische Störung, die die Produktion von Hämoglobin beeinflusst, was zu schweren Anämie und anderen Komplikationen führt. Während die Krankheit in Entwicklungsländern häufiger verbreitet ist, sehen die USA und Europa aufgrund der Bevölkerungsvielfalt zunehmende Fälle. Der Markt für Alpha-Thalassemia wird weitgehend von traditionellen Behandlungen wie Bluttransfusionen und Eisenchelierungstherapien angetrieben, aber die Entstehung neuer Therapien wie Mitapivat und Etavopivat bietet Hoffnung auf nicht-transfusionsabhängige Patienten. Diese neuen Therapien zielen darauf ab, die zugrunde liegenden genetischen Ursachen anzugehen und die Gesundheit der roten Blutkörperchen zu verbessern, was den Bedarf an häufigen Transfusionen deutlich verringern und die Lebensqualität verbessern könnte. Trotz des hohen Potenzials dieser Behandlungen bleiben Herausforderungen, darunter hohe Kosten, mangelndes Bewusstsein und eingeschränkter Zugang zu fortgeschrittenen Therapien in Entwicklungsregionen. Da Forschungsfortschritte und weitere Therapien auf den Markt kommen, wird die Alpha-Thalassemia-Landschaft mit besseren Ergebnissen für Patienten und einem signifikanten Wachstum auf dem Markt entwickelt.
Wettbewerbsübersicht von Alpha Thalassemia Markt
Zu den wichtigsten Akteuren des Alpha Thalassemia Market gehören Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc, Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc, Astrazeneca, F.Hoffman La Roche und Jans
Alpha Thalassemia Markt Marktführer
- Agios Pharmazeutika
- Novo Nordisk
- Bristol-Myers Squibb
- Forma Therapeuten
- Stille Therapie
Alpha Thalassemia Markt - Wettbewerbsrivalität

Alpha Thalassemia Markt
(Von großen Akteuren dominiert)
(Hoher Wettbewerb mit vielen Akteuren.)
Neueste Entwicklungen in Alpha Thalassemia Markt
- Im Januar 2024 kündigte Agios Pharmaceuticals positive Phase-III-Studienergebnisse für Mitapivat bei nicht-transfusionsabhängigen Alpha-Thalassemia-Patienten an, die eine signifikante Zunahme der Hämoglobinspiegel zeigten. Die Regelung wird im Jahr 2025 erwartet.
- Im September 2022 erwarb Novo Nordisk Forma Therapeutics und entwickelt jetzt Etavopivat, ein Untersuchungsmedikament, das die Notwendigkeit von Transfusionen durch Verbesserung der roten Blutkörperchengesundheit bei Alpha-Thalassemia-Patienten reduzieren soll.
Alpha Thalassemia Markt Segmentierung
- Nach Art der Krankheit
- Transfusion-Dependent Alpha Thalassemia (TDT)
- Nicht-Transfusion Dependent Alpha Thalassemia (NTDT)
- nach Behandlungsart
- Eisenkiesel Therapie
- Bluttransfusionen
- Knochen Marrow Transplantate
- Von Endbenutzer
- Pediatrie
- Erwachsene

Möchten Sie die Möglichkeit erkunden, einzelne Abschnitte dieses Berichts zu kaufen?
Häufig gestellte Fragen :
Wie groß ist der Alpha-Thalassemia-Markt?
Der Markt für Global Alpha Thalassemia wird im Jahr 2024 auf 4,4 Mrd. USD geschätzt und soll bis 2031 USD 9,1 Mrd. erreichen.
Was wird das CAGR des Alpha-Thalassemia-Marktes sein?
Die CAGR der Alpha-Thalassämie Der Markt wird von 2024-2031 auf 9,10% projiziert.
Was sind die wichtigsten Faktoren, die das Wachstum des Alpha-Thalassemia-Marktes behindern?
Der Mangel an zugelassenen Therapien für die Alpha-Talassämie, bei Patienten, die sich auf traditionelle Behandlungen wie Bluttransfusionen und Eisenchelierung und hohe Kosten und begrenzten Zugang zu fortgeschrittenen Behandlungen verlassen, vor allem in Entwicklungsländern, wo die Prävalenz höher ist, sind die wichtigsten Faktoren, die das Wachstum des Alpha-Thalassemia-Marktes behindern.
Was sind die wichtigsten Faktoren, die das Alpha-Thalassemia-Marktwachstum vorantreiben?
Das zunehmende Bewusstsein und die Forschung an genetischen Störungen wie Alpha-Talassämie, die zu früherer Diagnose und verbesserten Behandlungsansätzen und vielversprechenden Pipeline-Therapien wie Mitapivat, Etavopivat und Luspatercept führen, werden erwartet, den Markt zu treiben, insbesondere für nicht-transfusionsabhängige Fälle sind die wichtigsten Faktoren, die den Alpha-Thalassemia-Markt treiben.
Welches ist die führende Art von Krankheiten im Alpha-Thalassemia-Markt?
Nicht-Transfusion Dependent Alpha Thalassemia (NTDT) ist die führende Art von Krankheitssegment.
Welche sind die wichtigsten Akteure im Alpha-Thalassemia-Markt?
Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc, Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc, Astrazeneca, F.Hoffman La Roche, Jansen Pharmaceutical sind die wichtigsten Spieler.