Alpha Thalassémie Marché ANALYSE DE LA TAILLE ET DU PARTAGE - TENDANCES DE CROISSANCE ET PRÉVISIONS (2024 - 2031)
Alpha Thalassémie Le marché est segmenté par type de maladie (Thalassémie alpha transfusionnelle (TDT), Thalassémie alpha non transfusionnelle (NTDT),....
Alpha Thalassémie Marché Taille
Taille du marché en USD Bn
TCAC9.10%
| Période d'étude | 2024 - 2031 |
| TCAC | 9.10% |
| Concentration du marché | Medium |
| Principaux acteurs | Agios Pharmaceutiques, Novo Nordisk, Bristol-Myers Squibb, Forme thérapeutique, Silence thérapeutique et parmi d'autres |
Merci de nous le faire savoir !
Alpha Thalassémie Marché Analyse
Le marché mondial de la thalassémie alpha est estimé à 4,4 milliards de dollars en 2024 et devrait atteindre 9,1 milliards de dollars par 2031, en croissance à un taux de croissance annuel composé (TCAC) de 9,10 % de 2024 à 2031. Alpha Thalassemia est un trouble sanguin causé par des mutations des gènes HBA1 et HBA2. La prévalence croissante des troubles de l'alpha-thalassémie dans le monde devrait stimuler la croissance du marché pendant la période de prévision.
Le marché connaît une croissance positive en raison de la prise de conscience croissante du trouble et de ses options de traitement. De plus, de nouveaux produits pour traiter les anomalies génétiques sous-jacentes et des programmes de dépistage améliorés aident à détecter les cas contribuent à l'expansion du marché. Cependant, l'absence d'options de traitement symptomatiques et l'absence de médicaments modifiant la maladie sont quelques-uns des défis qui freinent la croissance du marché.
Alpha Thalassémie Marché Tendances
Pilote du marché - Sensibilisation accrue et recherche sur les troubles génétiques tels que l'alpha Thalassémie, menant à un diagnostic plus précoce et à des approches de traitement améliorées.
Au cours de la dernière décennie, il y a eu une augmentation substantielle des initiatives de sensibilisation entreprises à l'échelle mondiale pour sensibiliser le public aux troubles génétiques du sang comme la thalassémie alpha. Plusieurs organisations à but non lucratif et groupes de défense des patients ont travaillé avec diligence pour éduquer la population en général ainsi que les fournisseurs de soins de santé sur les signes, les symptômes et les options de diagnostic et de traitement disponibles pour ces maladies héréditaires. De plus, avec l'aide financière du gouvernement et du secteur privé, de vastes activités de recherche sont menées par des établissements universitaires et des sociétés pharmaceutiques pour approfondir les connaissances cliniques sur la physiopathologie et les antécédents naturels de ces maladies rares. Cela a permis de mieux comprendre le trouble et de le détecter rapidement.
Des programmes de dépistage systématique ont été mis en place dans de nombreux pays pour tester les femmes enceintes et les nouveau-nés en cas d'hémoglobines courantes. On dispose également de plus en plus d'options de dépistage et de diagnostic prénatals pour aider les mères enceintes à évaluer le risque et à se préparer aux défis que pose la prise en charge d'un enfant touché. Ensemble, ces efforts ont mené à un plus grand nombre de cas de thalassémie alpha diagnostiqués durant le stade foetal ou la petite enfance elle-même que dans le passé, alors que la plupart des patients resteraient non diagnostiqués pendant des années. Le diagnostic en temps opportun permet des soins et des interventions adaptés dès le début pour soulager les symptômes et gérer correctement l'état. Il contribue également à réduire les complications et les effets sur la santé à long terme. Tous ces facteurs liés à la croissance de la recherche et aux nouvelles connaissances contribuent positivement à des taux de diagnostic plus élevés et à des voies de soins optimisées pour les patients atteints de thalassémie alpha.
Pilote de marché - Thérapies de pipeline prometteuses pour conduire le marché
Le marché du traitement par la thalassémie alpha devrait connaître des possibilités de croissance lucratives au cours des prochaines années, sous l'effet d'un afflux de nouveaux candidats au traitement actuellement en développement. Plusieurs sociétés pharmaceutiques et start-ups en biotechnologie ont reconnu qu'il s'agissait d'un domaine dans lequel les besoins ne sont pas satisfaits et travaillent activement à des thérapies susceptibles de modifier les maladies par divers mécanismes d'action. Si elles sont traduites avec succès en médicaments approuvés, ces options de traitement innovatrices peuvent transformer les paradigmes de soins existants, en particulier pour les cas d'alpha Thalassémie non dépendants de la transfusion.
Parmi les thérapies pipelinières les plus avancées qui devraient générer des revenus importants, mentionnons Mitapivat par Agios, Etavopivat par Protagonist Therapeutics et Luspatercept par Acceleron/Celgene. Mitapivat est un activateur oral de petite molécule pyruvate kinase de première classe qui a montré des résultats prometteurs dans le traitement efficace du dysfonctionnement sous-jacent des globules rouges dans les études de phase II et est actuellement dans l'essai de phase III. De même, l'Etavopivat, un autre activateur de pyruvate kinase par voie orale, a montré une réduction de l'hémolyse extravasculaire au cours des premiers essais. Luspatercept, un agent de maturation érythroïde expérimental au stade avancé, a terminé la phase III avec succès et pourrait être le premier traitement approuvé par la FDA spécifiquement destiné aux patients de la thalassémie non dépendante de la transfusion.
S'ils sont approuvés, ces nouveaux agents peuvent offrir aux patients une option de traitement oral pour réduire significativement les transfusions et construire des globules rouges plus sains sans visites régulières à l'hôpital pour le soutien transfusionnel. Ils aideront également à répondre à un besoin non satisfait pour le traitement du sous-ensemble de patients non transfusionnels qui jusqu'à présent avaient des options thérapeutiques approuvées minimales disponibles. On s'attend à ce que ces progrès annoncent une nouvelle ère dans la gestion de la thalassémie Alpha au cours des cinq prochaines années et au-delà.
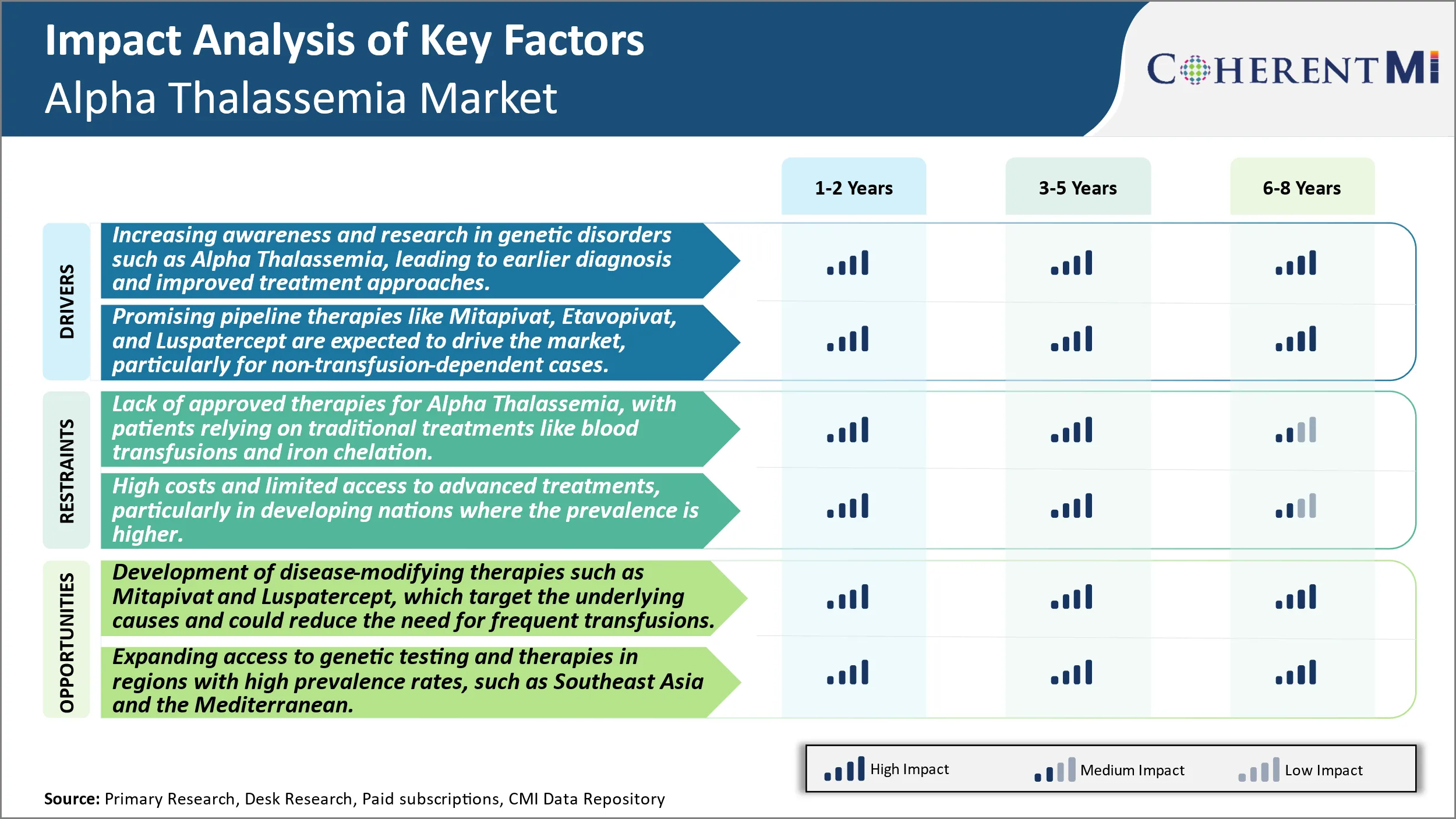
Défi du marché - Absence de thérapies approuvées pour l'alpha Thalassémie, les patients se fondant sur des traitements traditionnels comme les transfusions sanguines et la chélation du fer.
L'absence de thérapies de modification de la maladie approuvées pour la thalassémie alpha pose un défi important aux patients et à l'industrie. Les patients dépendent actuellement de mesures de soutien comme des transfusions sanguines régulières et la chélation du fer pour gérer les symptômes d'anémie sévère. Cependant, ces traitements traditionnels ne sont pas un remède et nécessitent une adhérence à vie qui peut affecter la qualité de vie. Les transfusions sanguines comportent des risques de surcharge en fer à long terme si elles ne sont pas gérées correctement par chélation. La nécessité de surveiller régulièrement les niveaux de fer et d'administrer la chélation Par voie intraveineuse ou orale, le traitement impose une charge aux patients. Du point de vue de l'industrie, l'absence d'options thérapeutiques curatives ou modifiant les maladies limite le potentiel de croissance. Les entreprises pharmaceutiques ont moins d'incitations à investir massivement dans la recherche et le développement de médicaments pour une maladie rare, avec seulement des options de soins de soutien disponibles. Dans l'ensemble, les besoins médicaux non satisfaits d'un traitement stimulent l'intérêt des patients et de l'industrie pour de nouvelles thérapies qui peuvent cibler la pathologie sous-jacente.
Opportunité de marché : Développement de thérapies modifiant la maladie pour créer de nouvelles possibilités.
Il existe des possibilités prometteuses dans le développement de thérapies modifiant la maladie pour la thalassémie alpha qui ciblent la cause profonde de la production inefficace d'hémoglobine. Deux médicaments, Mitapivat et Luspatercept, font l'objet d'essais cliniques en fin de cycle pour étudier leur potentiel de réduction de la dépendance transfusionnelle. Mitapivat active la pyruvate kinase qui pourrait rétablir la santé et la fonction des globules rouges. Dans les études cliniques réalisées jusqu'à présent, il a démontré une réduction de la charge transfusionnelle chez certains patients. Luspatercept agit en liant et en inhibant les protéines responsables de l' érythropoïèse inefficace, permettant ainsi une maturation normale des globules rouges. Son approbation pour la bêta-thalassémie a jeté les bases d'études sur la thalassémie alpha. Si ces thérapies réussissent, elles peuvent aider à améliorer les résultats cliniques en permettant de réduire le nombre de transfusions et de réduire la gestion de la surcharge en fer à long terme. Leur approbation pourrait revigorer le marché en répondant au besoin critique de traitements curatifs ou modifiant la maladie pour les patients atteints de thalassémie alpha.
Préférences des prescripteurs de Alpha Thalassémie Marché
La thalassémie alpha est généralement traitée selon deux grandes lignes de traitement - la première et la seconde. Dans le traitement de première intention, la plupart des prescripteurs préfèrent l'hydroxyurée dans les cas légers à modérés. Hydroxyurée, commercialisée sous les marques Hydrea et Droxia, vise à réduire la prolifération anormale de l'hémoglobine et à gérer les symptômes d'anémie.
Pour les cas plus graves nécessitant des transfusions sanguines régulières, les prescripteurs choisissent généralement le traitement chélateur en plus des transfusions. Le déférasirox (Exjade) et la défériprone (Ferriprox) sont des options de choix pour le chélateur de fer prescrites pour prévenir l'hémochromatose causée par l'absorption excessive de fer par les transfusions régulières.
Si un patient ne réagit pas adéquatement ou devient indépendant de la transfusion, les prescripteurs envisagent des traitements de seconde intention. Une greffe de moelle osseuse est recommandée aux patients admissibles pour traiter définitivement la maladie. Toutefois, en raison des risques encourus, les prescripteurs évaluent soigneusement l'aptitude à la transplantation en fonction de l'âge, du stade de la maladie et de la disponibilité d'un donneur équivalent.
Pour les patients non admissibles à la transplantation, les prescripteurs prescrivent Luspatercept-aamt (Reblozyl), un agent de maturation érythroïde nouvellement approuvé, en association avec les meilleurs soins de soutien, y compris les transfusions et la chélation du fer. Reblozyl peut réduire la dépendance transfusionnelle chez certains patients.
Dans l'ensemble, les prescripteurs recherchent des traitements qui soulagent les symptômes d'anémie, réduisent les besoins transfusionnels et tiennent compte de facteurs individuels comme la gravité, le taux de progression et la réponse aux traitements antérieurs. Cela aide à déterminer la ligne et le schéma de traitement le plus approprié pour chaque cas de thalassémie alpha.
Analyse des options de traitement de Alpha Thalassémie Marché
La thalassémie alpha présente différents niveaux de sévérité selon le nombre de gènes alpha-globins défectueux. Les plans de traitement sont adaptés en fonction du type spécifique. Sous des formes légères, aucun traitement ne peut être nécessaire car les patients sont souvent asymptomatiques. Sous des formes modérées, la première ligne de traitement est la supplémentation en acide folique, qui contribue à favoriser la production de globules rouges. Pour les formes sévères, des transfusions sanguines régulières sont nécessaires pour gérer les symptômes de l'anémie. Le traitement préféré est la transfusion de globules rouges toutes les 2 à 5 semaines selon l'individu. Cela aide à réduire d'autres complications en maintenant un taux constant d'hémoglobine. Le traitement par la chilation par la déféroxamine est également administré pour éliminer l'excès de fer des transfusions précédentes. Pour le type le plus grave appelé maladie HbH, la greffe de moelle osseuse est la seule option curative. Cependant, il est livré avec des risques et une correspondance parfaite des donneurs est nécessaire. La transplantation remplace la moelle osseuse défectueuse par des cellules donneurs saines. Des médicaments comme le busulfan et le cyclophosphamide sont fournis comme conditionnement pré-transplanté pour éliminer les cellules défectueuses.
Pour les patients qui ne peuvent pas être transplantés, la prise en charge se concentre sur les transfusions sanguines tout au long de la vie ainsi que sur le traitement par chélation afin d'éviter de graves dommages aux organes dus à la surcharge en fer. Une surveillance étroite aide à déterminer des horaires de transfusion optimaux adaptés à chaque patient. Dans l'ensemble, une approche multidisciplinaire des soins est nécessaire pour gérer efficacement l'état et améliorer la qualité de vie en fonction du niveau de gravité.
Stratégies gagnantes clés adoptées par les principaux acteurs de Alpha Thalassémie Marché
Adoption de Gene Traitement: Bluebird Bio a adopté une stratégie de thérapie génique pour Alpha Thalassemia en 2019. Ils ont développé LentiGlobin, un traitement de thérapie génique ciblant le défaut génétique sous-jacent qui cause Alpha Thalassémie. Au cours des essais cliniques, LentiGlobin a démontré une indépendance transfusionnelle significative chez les patients, éliminant ou réduisant leur dépendance aux transfusions sanguines régulières. Cette approche innovante de thérapie génique a transformé le paysage de traitement de la thalassémie alpha. Elle a été approuvée en Europe en 2019, ce qui en a fait la première thérapie génique approuvée pour la maladie.
Stratégie d'acquisition : Celgene a poursuivi une stratégie d'acquisition en achetant Agios Pharmaceuticals en 2018 pour USD7 bn. Agios avait un médicament expérimental appelé mitapivat dans les essais de phase 3 pour la thalassémie. Cela a permis à Celgene d'accéder à un oléoduc prometteur pour Alpha Thalassemia. En 2020, mitapivat a été approuvé par la FDA pour la thalassémie, devenant le premier traitement de modification de la maladie approuvé pour réduire le fardeau transfusionnel pour les populations de patients non-bêta thalassémie.
Collaborations et partenariats : Global Blood Therapeutics s'est associé à des pharmacies spécialisées et à des organismes sans but lucratif axés sur les maladies rares pour élargir l'accès à leur produit approuvé par la FDA Oxbryta pour le SCD. Ils ont fourni une aide financière et des programmes d'aide co-payés aux patients admissibles en thalassémie alpha. Cela a aidé à surmonter les obstacles à l'abordabilité et à stimuler l'absorption d'Oxbryta, ce qui réduit l'anémie et la dépendance à l'égard des transfusions sanguines. Les prescriptions pour Oxbryta dans l'indice Alpha Thalassémie ont doublé en 2021 par rapport à 2020.
Ces exemples démontrent des stratégies réussies comme l'innovation en thérapie génique, des acquisitions ciblées et des partenariats pour élargir l'accès et le remboursement.
Analyse segmentaire de Alpha Thalassémie Marché
Points de vue, par type de maladie : Le besoin de croissance pour la transfusion-dépendant Alpha Thalassémie (TDT) est évident.
Selon le type de maladie, l'Alpha Thalassémie (TDT) transfusionnelle devrait contribuer 52,5 % en 2024 en raison de la nature critique des soins requis par les patients. La TDT est une forme sévère qui nécessite des transfusions sanguines régulières à vie pour survivre. Les patients présentent une anémie sévère et d'autres complications si les transfusions ne sont pas correctement gérées. Cela rend le traitement non négociable et une priorité de soins de santé primaires. Les familles de patients atteints de TDT sont confrontées à d'immenses fardeaux médicaux et financiers pour maintenir des thérapies transfusionnelles à long terme. Les problèmes psychosociaux sont également fréquents en raison de la dépendance aux services médicaux. Par conséquent, les systèmes de soins de santé sont incités à optimiser l'accès au sang assorti de qualité et à investir dans des programmes de transfusion pour prévenir la mortalité.

Insights, par type de traitement, la thérapie par chélation du fer est prête à enregistrer une croissance remarquable dans les années à venir.
Par type de traitement, Chélation de fer La thérapie devrait contribuer à hauteur de 36,1 % en 2024 en raison de son adoption généralisée comme intervention de première ligne. Les transfusions s'attaquent efficacement à l'anémie mais introduisent la surcharge en fer comme un effet secondaire inévitable si elle n'est pas gérée. Le traitement par la chélation vise à lier et à éliminer l'excès de fer accumulé dans les organes vitaux par le biais de doses quotidiennes multiples. La desferrioxamine par perfusion sous-cutanée reste un traitement standard à l'échelle mondiale malgré ses inconvénients. Les nouveaux agents oraux comme la défériprone et le déférasirox ont gagné en préférence en raison d'une meilleure conformité de l'administration orale. Alors que l'espérance de vie des patients atteints de TDT augmente avec des soins de qualité, le contrôle à long terme du fer est important pour éviter les complications et maximiser le bien-être.
Insights, par utilisateur final, la pédiatrie devrait connaître une croissance remarquable au cours de la période de prévision.
Par l'utilisateur final, la pédiatrie représente la part la plus élevée du marché en raison de la TDT souvent diagnostiquée à la naissance ou dans la petite enfance. Les symptômes se manifestent dès l'enfance, nécessitant une intervention rapide. L'utilisation des soins de santé est intensive, depuis les évaluations de dépistage et de diagnostic jusqu'aux thérapies transfusionnelles longitudinales tout au long du développement. Outre les besoins médicaux, les patients pédiatriques ont également besoin d'une vaste formation, de conseils et d'un soutien multidisciplinaire coordonné pour assurer leur respect. La gestion en temps opportun au milieu des étapes de croissance vise à optimiser les jalons physiques, cognitifs et sociaux. Bien que les soins aux adultes demeurent essentiels, la demande pédiatrique a priorité pour traiter la maladie dès son apparition et minimiser les conséquences à long terme.
Informations supplémentaires sur Alpha Thalassémie Marché
Alpha Thalassemia est un trouble génétique qui affecte la production d'hémoglobine, entraînant une anémie sévère et d'autres complications. Si la maladie est plus répandue dans les pays en développement, les États-Unis et l'Europe voient des cas croissants en raison de la diversité de la population. Le marché de l'Alpha Thalassemia est largement alimenté par des traitements traditionnels tels que les transfusions sanguines et les thérapies de chélation de fer, mais l'émergence de nouvelles thérapies comme Mitapivat et Etavopivat offre de l'espoir aux patients non dépendants de la transfusion. Ces nouvelles thérapies visent à s'attaquer aux causes génétiques sous-jacentes et à améliorer la santé des globules rouges, ce qui pourrait réduire considérablement le besoin de transfusions fréquentes et améliorer la qualité de vie. Malgré le potentiel élevé de ces traitements, des défis subsistent, notamment des coûts élevés, un manque de sensibilisation et un accès limité aux thérapies de pointe dans les régions en développement. Au fur et à mesure que la recherche progresse et que d'autres thérapies arrivent sur le marché, le paysage Alpha Thalassemia devrait évoluer, avec de meilleurs résultats pour les patients et une croissance significative du marché.
Aperçu concurrentiel de Alpha Thalassémie Marché
Les principaux acteurs du marché Alpha Thalassemia sont Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc., Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc., Astrazeneca, F.Hoffman La Roche et Jansen Pharmaceutical.
Alpha Thalassémie Marché Leaders
- Agios Pharmaceutiques
- Novo Nordisk
- Bristol-Myers Squibb
- Forme thérapeutique
- Silence thérapeutique
Alpha Thalassémie Marché - Rivalité concurrentielle

Alpha Thalassémie Marché
(Dominé par des acteurs majeurs)
(Très compétitif avec de nombreux acteurs.)
Développements récents dans Alpha Thalassémie Marché
- En janvier 2024, Agios Pharmaceuticals a annoncé des résultats positifs d'essais de phase III pour Mitapivat chez des patients d'Alpha Thalassémie non dépendants de la transfusion, montrant une augmentation significative des taux d'hémoglobine. L'approbation réglementaire est prévue en 2025.
- En septembre 2022, Novo Nordisk a acquis Forma Therapeutics et développe actuellement Etavopivat, un médicament oral expérimental qui vise à réduire le besoin de transfusions en améliorant la santé des globules rouges chez les patients atteints d'alpha Thalassémie.
Alpha Thalassémie Marché Segmentation
- Par type de maladie
- Dépendant de la transfusion Alpha Thalassémie (TDT)
- Alpha Thalassémie (NTDT) non dépendante de la transfusion
- Par type de traitement
- Chélation de fer Thérapie
- Transfusions sanguines
- Transplantations de moelle osseuse
- Par utilisateur final
- Pédiatrie
- Adulte

Souhaitez-vous explorer l'option d'achat sections individuelles de ce rapport ?
Questions fréquemment posées :
Quelle est la taille du marché de la thalassémie Alpha?
Le marché mondial de la thalassémie alpha est estimé à 4,4 milliards de dollars en 2024 et devrait atteindre 9,1 milliards de dollars en 2031.
Quel sera le TCAC du marché de la thalassémie alpha?
Le TCAC de la thalassémie alpha Le marché devrait passer de 2024 à 2031 à 9,10 %.
Quels sont les principaux facteurs qui entravent la croissance du marché de la thalassémie alpha?
L'absence de thérapies approuvées pour la thalassémie alpha, les patients comptant sur des traitements traditionnels comme les transfusions sanguines et la chélation du fer et les coûts élevés et l'accès limité aux traitements avancés, en particulier dans les pays en développement où la prévalence est plus élevée sont les principaux facteurs qui entravent la croissance du marché de la thalassémie alpha.
Quels sont les principaux facteurs à l'origine de la croissance du marché de la thalassémie alpha?
La sensibilisation et la recherche croissantes dans le domaine des troubles génétiques tels que la thalassémie alpha, qui ont conduit à un diagnostic plus précoce et à des approches thérapeutiques améliorées et à des thérapies prometteuses comme Mitapivat, Etavopivat et Luspatercept, sont censées stimuler le marché, en particulier pour les cas non dépendants de la transfusion, sont les principaux facteurs à l'origine du marché de la thalassémie alpha.
Quel est le principal type de maladie sur le marché de la thalassémie alpha?
Alpha Thalassemia (NTDT) est le premier segment de type de maladie dépendant de la transfusion.
Quels sont les principaux acteurs du marché Alpha Thalassemia ?
Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc., Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc., Astrazeneca, F.Hoffman La Roche, Jansen Pharmaceutical sont les principaux acteurs.