Alpha Thalassemia Mercado ANÁLISE DE TAMANHO E PARTICIPAÇÃO - TENDÊNCIAS DE CRESCIMENTO E PREVISÕES (2024 - 2031)
Alpha Thalassemia O mercado é segmentado por tipo de doença (Transfusion-Dependent Alpha Thalassemia (TDT), não-Transfusion Dependente Alpha Thalassem....
Alpha Thalassemia Mercado Tamanho
Tamanho do mercado em USD Bn
CAGR9.10%
| Período de estudo | 2024 - 2031 |
| CAGR | 9.10% |
| Concentração de Mercado | Medium |
| Principais jogadores | Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Terapêutica, Terapêutica de Silêncio e entre outros |
por favor, avise-nos!
Alpha Thalassemia Mercado Análise
Estima-se que o mercado Global Alpha Thalassemia seja avaliado em USD 4.4 bn em 2024 e é esperado alcançar USD 9.1 bn por 2031, crescendo a uma taxa de crescimento anual composta (CAGR) de 9,10% de 2024 a 2031. Alpha Thalassemia é um distúrbio sanguíneo causado por mutações nos genes HBA1 e HBA2. Espera-se que a prevalência crescente de transtornos alfa-telassemia globalmente aumente o crescimento do mercado durante o período de previsão.
O mercado está testemunhando um crescimento positivo devido ao aumento da consciência sobre o transtorno e suas opções de tratamento. Além disso, novos desenvolvimentos de produtos para tratar as anormalidades genéticas subjacentes e programas de rastreamento aprimorados ajudam a detectar casos estão contribuindo para a expansão do mercado. No entanto, a ausência de opções de tratamento sintomático e a falta de medicamentos modificadores de doenças são alguns dos desafios que restringem o crescimento do mercado.
Alpha Thalassemia Mercado Tendências
Driver de mercado - Aumentando a conscientização e a pesquisa em distúrbios genéticos, como Alpha Thalassemia, levando ao diagnóstico anterior e melhores abordagens de tratamento.
Ao longo da última década houve um aumento substancial nas iniciativas de conscientização realizadas globalmente para espalhar a conscientização sobre os transtornos genéticos do sangue como Alpha Thalassemia. Várias organizações sem fins lucrativos e grupos de defesa do paciente têm trabalhado diligentemente para educar a população geral, bem como prestadores de cuidados de saúde sobre os sinais, sintomas e opções de diagnóstico e tratamento disponíveis para tais condições herdadas. Além disso, apoiado pelo governo e financiamento privado, extensas atividades de pesquisa estão sendo realizadas por instituições acadêmicas e empresas farmacêuticas para obter insights clínicos mais profundos sobre a fisiopatologia e história natural dessas doenças raras. Isso melhorou significativamente a compreensão sobre o transtorno e ajudou na detecção precoce.
Programas de rastreamento de rotina foram implementados em muitos países para testar mulheres grávidas e recém-nascidos para hemoglobinopatias comuns. As opções de rastreamento e diagnóstico pré-natal também estão cada vez mais disponíveis para ajudar as mães grávidas a avaliar o risco e preparar os desafios de cuidar de uma criança afetada. Juntos, esses esforços levaram a mais casos de Thalassemia Alfa sendo diagnosticados durante a fase fetal ou a própria infância em comparação com o passado quando a maioria dos pacientes permaneceria não diagnosticada por anos. O diagnóstico oportuna permite cuidados e intervenções sob medida do início para aliviar os sintomas e gerenciar a condição corretamente. Também ajuda a reduzir complicações e impactos de saúde no longo prazo. Todos esses fatores relacionados ao crescimento da pesquisa e novos insights estão contribuindo positivamente para maiores taxas de diagnóstico e vias de atendimento otimizadas para pacientes da Thalassemia Alpha.
Driver de mercado - Promising Pipeline Therapies para dirigir o mercado
O mercado de tratamento Alpha Thalassemia é susceptível de ver oportunidades de crescimento lucrativo nos próximos anos impulsionado por um pipeline de inchaço de novos candidatos à droga atualmente em desenvolvimento. Várias empresas farmacêuticas e start-ups biotecnológicos reconheceram isso como uma área de necessidade não satisfeita e estão trabalhando ativamente em potenciais terapias modificadoras de doenças através de diversos mecanismos de ação. Se traduzidas com sucesso em medicamentos aprovados, essas opções de tratamento inovadoras têm a capacidade de transformar os paradigmas existentes de cuidado, particularmente para casos não-transfusion-dependentes de Alpha Thalassemia.
Algumas das terapias de pipeline mais avançadas que são esperados para gerar receitas significativas incluem Mitapivat por Agios, Etavopivat por Protagonist Therapeutics, e Luspatercept por Acceleron/Celgene. Mitapivat é um ativador pyruvate kinase pyruvate da molécula pequena oral, que mostrou resultados promissores no tratamento eficaz da disfunção da célula vermelha subjacente nos estudos da fase II e está atualmente no teste da fase III. Da mesma forma, Etavopivat, outro ativador pyruvate kinase oral, demonstrou redução na hemolise extravascular em ensaios iniciais. Luspatercept, um agente de maturação eritroide investigacional de última fase, concluiu a Fase III com sucesso e poderia ser a primeira terapia aprovada pela FDA especificamente destinada a pacientes não-transfusão-dependentes da Thalassemia.
Se aprovado, esses novos agentes podem oferecer aos pacientes uma opção de tratamento oral para reduzir significativamente as transfusões e construir células sanguíneas vermelhas mais saudáveis sem visitas hospitalares regulares para apoio à transfusão. Eles também ajudarão a abordar uma necessidade não satisfeita de tratar o subconjunto não-transfusão de pacientes que até agora tinham opções terapêuticas aprovadas mínimas disponíveis. Este progresso do pipeline é esperado para anunciar uma nova idade na gestão Alpha Thalassemia ao longo dos próximos cinco anos e além.
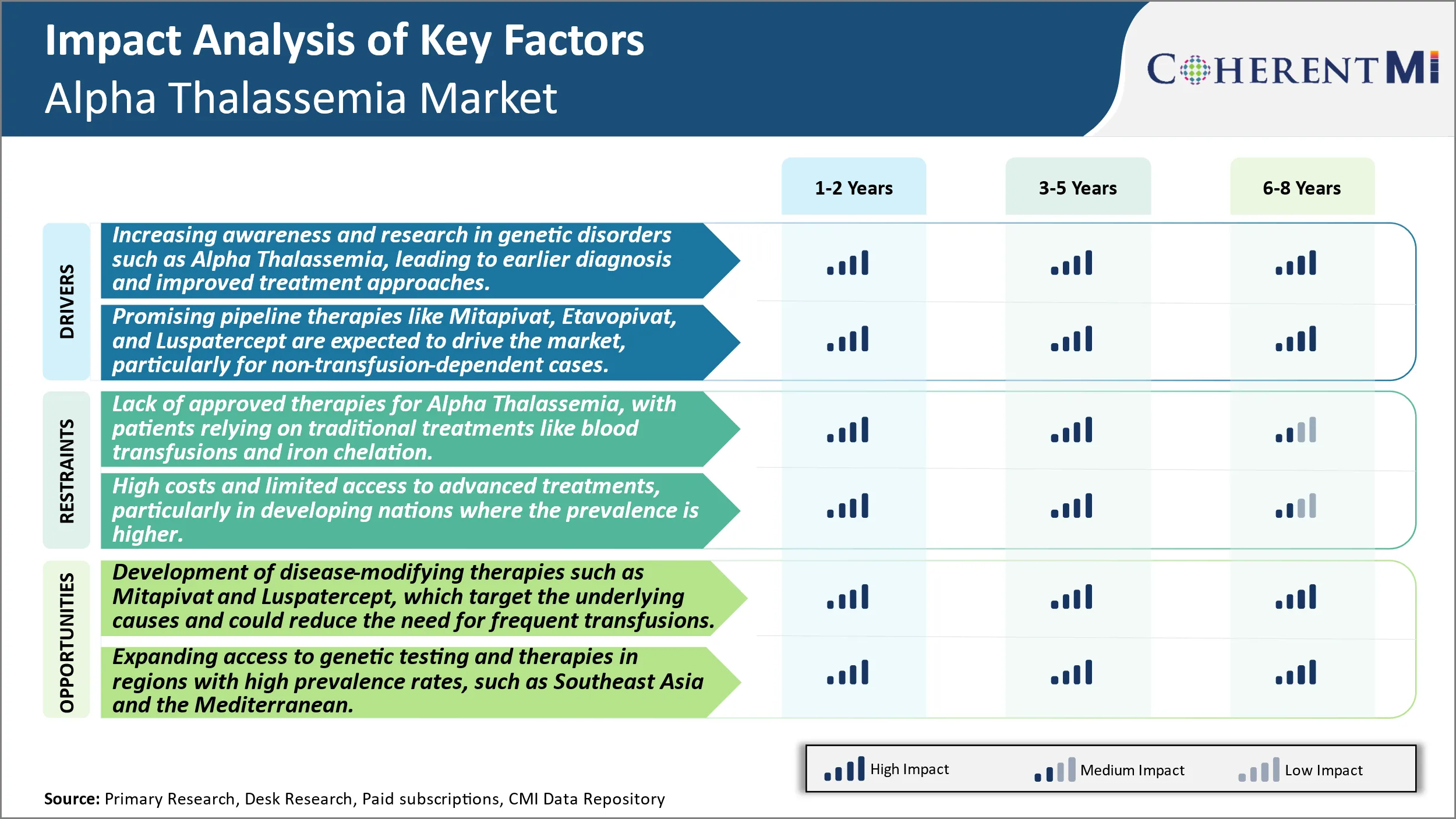
Desafio de Mercado - Falta de Terapias Aprovadas para a Thalassemia Alfa, Com Pacientes Baseando-se em Tratamentos Tradicionais Como Transfusões de Sangue e Chelation de Ferro.
A falta de terapias homologadas para a talassemia alfa representa um desafio significativo para ambos os pacientes e para a indústria. Atualmente, os pacientes dependem de medidas de apoio, como transfusões sanguíneas regulares e quilatação de ferro para gerenciar sintomas graves de anemia. No entanto, esses tratamentos tradicionais não são uma cura e exigem a adesão ao longo da vida que pode impactar a qualidade de vida. As transfusões de sangue carregam riscos de sobrecarga de ferro no longo prazo, se não forem geridas corretamente com a quilatação. As necessidades de controlo regular dos níveis de ferro e de gestão da quilatação Intravenamente ou por via oral coloca uma carga de tratamento em pacientes. De uma perspectiva da indústria, a falta de opções curativas ou de tratamento modificador de doenças limita o potencial de crescimento. As empresas farmacêuticas têm menos incentivos para investir fortemente na pesquisa e desenvolvimento de drogas para uma doença rara com apenas opções de cuidados de apoio disponíveis. Em geral, a necessidade médica não satisfeita de uma cura impulsiona o interesse do paciente e da indústria em terapias novas que podem segmentar a patologia subjacente.
Oportunidade de mercado: Desenvolvimento de terapias de transformação de doenças para criar oportunidades novárias.
Há uma oportunidade promissora no desenvolvimento de terapias modificadoras de doenças para a talassemia alfa que visam a causa raiz da produção de hemoglobina ineficaz. Duas drogas, Mitapivat e Luspatercept, estão em ensaios clínicos de última fase investigando seu potencial para reduzir a dependência de transfusão. Mitapivat ativa a pyruvate kinase que poderia restaurar a saúde e função da célula vermelha do sangue. Em estudos clínicos até agora, demonstrou reduções na carga de transfusão para certos pacientes. Luspatercept funciona através da ligação e inibição de proteínas responsáveis pela eritropoiese ineficaz, permitindo assim a maturação normal das células vermelhas do sangue. Sua aprovação para beta-talassemia estabeleceu a base para estudos na talassemia alfa. Se bem sucedido, essas terapias podem ajudar a melhorar os resultados clínicos, permitindo menos transfusões e reduzindo o gerenciamento de sobrecarga de ferro ao longo do longo prazo. Sua aprovação poderia revigorar o mercado, cumprindo a necessidade crítica de tratamentos curativos ou modificadores de doenças para pacientes alfa talassemia.
Preferências dos prescritores de Alpha Thalassemia Mercado
A talassemia alfa é tipicamente tratada ao longo de duas linhas principais de tratamento - primeira linha e segunda linha. No tratamento de primeira linha, a maioria dos prescritores preferem hidroxiureia para casos leves a moderados. Hydroxyurea, comercializada sob nomes de marcas Hydrea e Droxia, trabalha para reduzir a proliferação de hemoglobina anormal e gerenciar sintomas de anemia.
Para casos mais graves que requerem transfusões de sangue regulares, os prescritores geralmente escolhem a terapia do quilator além de transfusões. Deferasirox (Exjade) e deferiprone (Ferriprox) são opções preferidas de chelator de ferro prescritas para evitar hemocromatose causada por excesso de absorção de ferro de transfusões regulares.
Se um paciente não responder adequadamente ou se tornar independente de transfusão, os prescritores consideram tratamentos de segunda linha. O transplante de medula óssea é recomendado para pacientes elegíveis para tratar definitivamente a doença. No entanto, devido aos riscos envolvidos, os prescritores avaliam cuidadosamente a adequação do transplante com base na idade, estágio de doença e disponibilidade de um doador combinado.
Para os pacientes não elegíveis para transplante, os prescritores prescrevem Luspatercept-aamt (Reblozyl), um agente de maturação eritroide recém-aprovado, em combinação com o melhor cuidado de suporte, incluindo transfusões e quilação de ferro. Reblozyl pode reduzir a dependência de transfusão em alguns pacientes.
Os prescritores globais buscam tratamentos que aliviam os sintomas da anemia, reduzem os requisitos de transfusão e consideram fatores individuais do paciente como gravidade, taxa de progressão e resposta às terapias anteriores. Isso ajuda a determinar a linha mais adequada e regime de tratamento para cada caso alfa talassemia.
Análise de Opção de Tratamento de Alpha Thalassemia Mercado
A talassemia alfa tem diferentes níveis de gravidade dependendo do número de genes de globin alfa defeituosos. Os planos de tratamento são adaptados com base no tipo específico. Em formas leves, nenhum tratamento pode ser necessário como os pacientes são frequentemente assintomáticos. Em formas moderadas, a primeira linha de tratamento é a suplementação de ácido fólico, o que ajuda a promover a produção de glóbulos vermelhos. Para formas graves, transfusões sanguíneas regulares são necessárias para gerenciar sintomas de anemia. O tratamento preferido é transfusões de glóbulos vermelhos a cada 2-5 semanas dependendo do indivíduo. Isso ajuda a reduzir outras complicações, mantendo níveis consistentes de hemoglobina. Terapia de quilatação usando deferoxamina também é dada para remover o excesso de ferro de transfusões anteriores. Para o tipo mais grave chamado doença HbH, o transplante de medula óssea é a única opção curativa. No entanto, ele vem com riscos e uma combinação perfeita doador é necessária. O transplante substitui a medula óssea com células doadoras saudáveis. Medicamentos como busulfan e ciclofosfamida são fornecidos como pré-transplante condicionado para eliminar células defeituosas.
Para pacientes que não podem ser submetidos a transplante, a gestão se concentra em transfusões de sangue ao longo da vida, juntamente com terapia de quilatação para evitar danos graves de órgãos de sobrecarga de ferro. Monitoramento próximo ajuda a determinar horários de transfusão ideais adaptados a cada paciente. No geral, uma abordagem multidisciplinar de cuidados é necessária para gerenciar efetivamente a condição e melhorar a qualidade de vida com base no nível de gravidade.
Principais estratégias vencedoras adotadas pelos principais participantes de Alpha Thalassemia Mercado
Adoção de Gene Terapia: Bluebird Bio adotou uma estratégia de terapia genética para Alpha Thalassemia em 2019. Eles desenvolveram LentiGlobin, um tratamento de terapia genética que visa o defeito genético subjacente que causa Alpha Thalassemia. Em ensaios clínicos, LentiGlobin demonstrou significativa independência de transfusão em pacientes, eliminando ou reduzindo sua dependência de transfusões de sangue regulares. Esta abordagem inovadora de terapia genética transformou a paisagem de tratamento para Alpha Thalassemia. Recebeu aprovação de marketing na Europa em 2019, tornando-se a primeira terapia genética aprovada para a doença.
Estratégia de Aquisição: Celgene prosseguiu uma estratégia de aquisição, adquirindo Agios Pharmaceuticals em 2018 para USD7 bn. Agios tinha uma droga experimental chamada mitapivat na fase 3 testes para a Thalassemia. Isso permitiu que Celgene ganhasse acesso a um conjunto promissor de pipeline de última fase para Alpha Thalassemia. Em 2020, a mitapivat foi aprovada pela FDA para a Thalassemia, tornando-se a primeira doença aprovada que modifica a terapia para reduzir a carga de transfusão para as populações de pacientes não beta talassemia.
Colaborações e parcerias: Global Blood Therapeutics em parceria com especialidades farmacêuticas e sem fins lucrativos focadas em doenças raras para expandir o acesso ao seu produto aprovado pela FDA Oxbryta para SCD. Eles forneceram ajuda financeira e programas de assistência co-pay para pacientes elegíveis Alpha Thalassemia. Isso ajudou a superar barreiras de acessibilidade e aumentar o consumo de Oxbryta, o que reduz a anemia e a dependência de transfusões de sangue. As prescrições para Oxbryta na indicação Alpha Thalassemia duplicaram em 2021 em comparação com 2020.
Esses exemplos demonstram estratégias bem sucedidas, como inovação em terapia genética, aquisições direcionadas e parcerias para ampliar o acesso e o reembolso.
Análise Segmental de Alpha Thalassemia Mercado
Insights, por tipo de doença: A necessidade crescente de transfusão-dependente Alpha Thalassemia (TDT) é evidente.
Por tipo de doença, o Transfusion-Dependent Alpha Thalassemia (TDT) deve contribuir 52,5% em 2024 devido à natureza crítica do cuidado exigido pelos pacientes. TDT é uma forma severa que requer transfusões de sangue regulares ao longo da vida para sobrevivência. Os pacientes experimentam anemia grave e outras complicações se as transfusões não forem adequadamente geridas. Isso torna o tratamento não negociável e uma prioridade primária à saúde. Famílias de pacientes TDT enfrentam enormes cargas médicas e financeiras para sustentar terapias de transfusão a longo prazo. Os desafios psicossociais também são prevalentes devido à dependência dos serviços médicos. Como resultado, os sistemas de saúde são solicitados a otimizar o acesso ao sangue combinado com qualidade e investir em programas de transfusão para prevenir a mortalidade.

Insights, por tipo de tratamento, a terapia de chelation de ferro é envenenada para registrar um crescimento notável nos anos vindouros.
Por tipo de tratamento, Chelation de ferro Espera-se que a terapia contribua 36,1% em 2024 devido à sua adoção generalizada como uma intervenção de linha de frente. As transfusões abordam efetivamente a anemia, mas introduzem a sobrecarga de ferro como um efeito lateral inevitável se deixada desmantelada. A terapia de quilatação visa ligar e remover o excesso de ferro acumulado em órgãos vitais através de vários horários de dose diária. A deferrioxamina por infusão subcutânea permanece um tratamento padrão globalmente, apesar de sua inconveniência. Agentes orais mais recentes como deferiprone e deferasirox ganharam preferência devido a melhor conformidade da administração oral. À medida que as expectativas de vida dos pacientes TDT aumentam com o cuidado de qualidade, o controle de ferro de longo prazo tem importância para evitar complicações e maximizar o bem-estar.
Insights, By End-user, Pediatric é esperado para testemunhar um crescimento notável no período de previsão.
Por usuário final, a Pediatria contribui com a maior parte do mercado devido ao TDT muitas vezes sendo diagnosticado no nascimento ou na infância. Os sintomas manifestam-se da infância, necessitando de intervenção rápida. A utilização de cuidados de saúde é intensiva desde a triagem e avaliações diagnósticas até terapias de transfusão longitudinal ao longo do desenvolvimento. Além dos requisitos médicos, os pacientes pediátricos também precisam de educação extensa, aconselhamento e apoio multidisciplinar coordenado para garantir a adesão. Gestão oportuna em meio a estágios de crescimento visa otimizar os marcos físicos, cognitivos e sociais. Enquanto o cuidado adulto permanece essencial, a demanda pediátrica tem precedência para abordar a doença desde o início e minimizar as consequências a longo prazo.
Informação adicional de Alpha Thalassemia Mercado
Alpha Thalassemia é um distúrbio genético que afeta a produção de hemoglobina, levando a anemia grave e outras complicações. Embora a doença seja mais prevalente nos países em desenvolvimento, os EUA e a Europa estão vendo casos crescentes devido à diversidade populacional. O mercado para Alpha Thalassemia é em grande parte impulsionado por tratamentos tradicionais, tais como transfusões de sangue e terapias de chelation de ferro, mas o surgimento de novas terapias como Mitapivat e Etavopivat oferece esperança para pacientes não-transfusion-dependentes. Estas novas terapias visam abordar as causas genéticas subjacentes e melhorar a saúde das células vermelhas do sangue, o que poderia reduzir significativamente a necessidade de transfusões frequentes e melhorar a qualidade de vida. Apesar do alto potencial desses tratamentos, os desafios permanecem, incluindo altos custos, falta de consciência e acesso limitado a terapias avançadas em regiões em desenvolvimento. À medida que a pesquisa progride e mais terapias atingem o mercado, espera-se que a paisagem Alpha Thalassemia evolua, com melhores resultados para pacientes e crescimento significativo no mercado.
Visão geral competitiva de Alpha Thalassemia Mercado
Os principais jogadores que operam no Mercado Alpha Thalassemia incluem Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc, Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc, Astrazeneca, F.Hoffman La Roche e Jansen Pharmaceutical.
Alpha Thalassemia Mercado Líderes
- Agios Pharmaceuticals
- Novo Nordisk
- Bristol-Myers Squibb
- Forma Terapêutica
- Terapêutica de Silêncio
Alpha Thalassemia Mercado - Rivalidade Competitiva

Alpha Thalassemia Mercado
(Dominado por grandes players)
(Altamente competitivo com muitos jogadores.)
Desenvolvimentos recentes em Alpha Thalassemia Mercado
- Em janeiro de 2024, a Agios Pharmaceuticals anunciou resultados positivos de teste para Mitapivat em pacientes não dependentes da transfusão Alpha Thalassemia, mostrando um aumento significativo nos níveis de hemoglobina. Aprovação regulatória é esperada em 2025.
- Em setembro de 2022, Novo Nordisk adquiriu Forma Therapeutics e agora está desenvolvendo Etavopivat, uma droga oral investigativa que visa reduzir a necessidade de transfusões, melhorando a saúde de glóbulos vermelhos em pacientes Alpha Thalassemia.
Alpha Thalassemia Mercado Segmentação
- Por tipo de doença
- Transfusion-Dependent Alpha Thalassemia (TDT)
- Não-Transfusão Dependente Alpha Thalassemia (NTDT)
- Por tipo de tratamento
- Chelation de ferro Terapia
- Transfusões de sangue
- Transplante de medula óssea
- Por usuário final
- Pediatras
- Adulto

Gostaria de explorar a opção de comprasecções individuais deste relatório?
Perguntas Frequentes :
Quão grande é o Mercado Alpha Thalassemia?
Estima-se que o mercado Global Alpha Thalassemia seja avaliado em USD 4.4 bn em 2024 e deverá chegar a USD 9.1 bn em 2031.
Qual será o CAGR do Mercado Alpha Thalassemia?
O CAGR da Thalassemia Alfa O mercado é projetado para ser 9,10% de 2024-2031.
Quais são os principais fatores que dificultam o crescimento do Mercado Alpha Thalassemia?
A falta de terapias aprovadas para a talassemia alfa, com pacientes que dependem de tratamentos tradicionais, como transfusões de sangue e quilatação de ferro e altos custos e acesso limitado a tratamentos avançados, particularmente em países em desenvolvimento, onde a prevalência é maior são os principais fatores que dificultam o crescimento do Mercado de Talassemia Alfa.
Quais são os principais fatores que impulsionam o crescimento do Mercado Alpha Thalassemia?
A crescente conscientização e pesquisa em distúrbios genéticos como a talassemia alfa, levando ao diagnóstico anterior e melhores abordagens de tratamento e terapias de pipeline promissoras como Mitapivat, Etavopivat, e Luspatercept são esperados para impulsionar o mercado, particularmente para casos não-transfusão dependentes são os principais fatores que impulsionam o Mercado de Talassemia Alfa.
Qual é o tipo de doença líder no mercado de Alpha Thalassemia?
Não-Transfusão Dependente Alpha Thalassemia (NTDT) é o principal segmento Tipo de Doença.
Quais são os principais jogadores que operam no Mercado Alpha Thalassemia?
Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc, Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc, Astrazeneca, F.Hoffman La Roche, Jansen Pharmaceutical são os principais jogadores.