Alpha Thalassemia Market SIZE AND SHARE ANALYSIS - GROWTH TRENDS AND FORECASTS (2024 - 2031)
Alpha Thalassemia Market is segmented By Type of Disease (Transfusion-Dependent Alpha Thalassemia (TDT), Non-Transfusion Dependent Alpha Thalassemia (....
Alpha Thalassemia Market Size
Market Size in USD Bn
CAGR9.10%
| Study Period | 2024 - 2031 |
| CAGR | 9.10% |
| Market Concentration | Medium |
| Major Players | Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics and Among Others. |
please let us know !
Alpha Thalassemia Market Analysis
The Global Alpha Thalassemia market is estimated to be valued at USD 4.4 bn in 2024 and is expected to reach USD 9.1 bn by 2031, growing at a compound annual growth rate (CAGR) of 9.10% from 2024 to 2031. Alpha Thalassemia is a blood disorder caused by mutations in the HBA1 and HBA2 genes. The increasing prevalence of alpha-thalassemia disorders globally is expected to drive the growth of the market during the forecast period.
The market is witnessing positive growth owing to rising awareness about the disorder and its treatment options. Furthermore, new product developments to treat the underlying genetic abnormalities and enhanced screening programs help detect cases are contributing to the market expansion. However, the absence of symptomatic treatment options and lack of disease-modifying drugs are some of the challenges restraining the market growth.
Alpha Thalassemia Market Trends
Market Driver - Increasing awareness and research in genetic disorders such as Alpha Thalassemia, leading to earlier diagnosis and improved treatment approaches.
Over the past decade there has been a substantial increase in awareness initiatives undertaken globally to spread awareness about genetic blood disorders like Alpha Thalassemia. Several non-profit organizations and patient advocacy groups have been working diligently towards educating the general population as well as healthcare providers about the signs, symptoms, and available diagnosis and treatment options for such inherited conditions. Moreover, supported by government and private funding, extensive research activities are being carried out by academic institutions and pharma companies to gain deeper clinical insights into the pathophysiology and natural history of these rare diseases. This has significantly improved understanding about the disorder and helped in early detection.
Routine screening programs have been implemented in many countries to test pregnant women and newborns for common hemoglobinopathies. Prenatal screening and diagnosis options are also increasingly available to help expectant mothers assess risk and prepare for the challenges of caring for an affected child. Together, these efforts have led to more Alpha Thalassemia cases being diagnosed during fetal stage or early childhood itself compared to past when most patients would remain undiagnosed for years. Timely diagnosis allows for tailored care and interventions from beginning to alleviate symptoms and manage the condition properly. It also helps reduce complications and health impacts in the long run. All of these factors related to growing research and new insights are positively contributing to higher diagnosis rates and optimized care pathways for Alpha Thalassemia patients.
Market Driver - Promising Pipeline Therapies to Drive the Market
The Alpha Thalassemia treatment market is likely to see lucrative growth opportunities in the coming years driven by a swelling pipeline of novel drug candidates currently under development. Multiple pharmaceutical companies and biotech start-ups have recognized this as an area of unmet need and are actively working on potential disease-modifying therapies through diverse mechanisms of action. If successfully translated into approved drugs, these innovative treatment options have the capability to transform the existing paradigms of care, particularly for non-transfusion-dependent cases of Alpha Thalassemia.
Some of the most advanced pipeline therapies which are expected to drive significant revenues include Mitapivat by Agios, Etavopivat by Protagonist Therapeutics, and Luspatercept by Acceleron/Celgene. Mitapivat is a first-in-class, oral small molecule pyruvate kinase activator which has shown promising results in effectively treating the underlying red blood cell dysfunction in Phase II studies and is currently in Phase III trial. Likewise, Etavopivat, another oral pyruvate kinase activator, has demonstrated reduction in extravascular hemolysis in early trials. Luspatercept, a late-stage investigational erythroid maturation agent, has completed Phase III successfuly and could be the first FDA approved therapy specifically intended for non-transfusion-dependent Thalassemia patients.
If approved, these novel agents may offer patients an oral treatment option to significantly reduce transfusions and build healthier red blood cells without regular hospital visits for transfusion support. They will also help address an unmet need for treating the non-transfusion subset of patients who until now had minimal approved therapeutic options available. This pipeline progress is expected to herald a new age in Alpha Thalassemia management over the next five years and beyond.
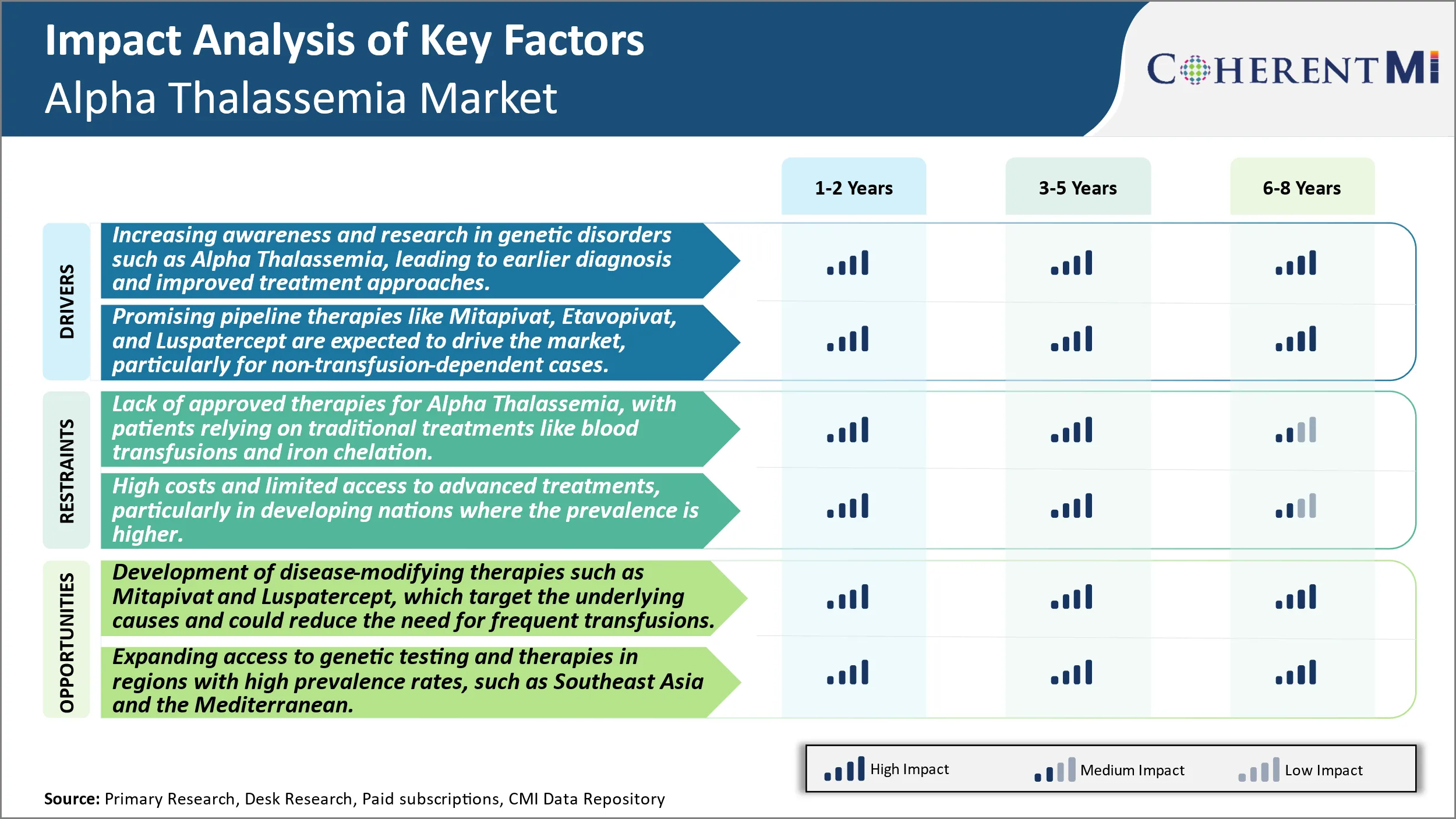
Market Challenge - Lack of Approved Therapies for Alpha Thalassemia, With Patients Relying on Traditional Treatments Like Blood Transfusions and Iron Chelation.
The lack of approved disease-modifying therapies for alpha thalassemia poses a significant challenge for both patients and the industry. Patients are currently reliant on supportive measures like regular blood transfusions and iron chelation to manage severe anemia symptoms. However, these traditional treatments are not a cure and require life-long adherence which can impact quality of life. Blood transfusions carry risks of iron overload in the long run if not managed properly with chelation. The needs of regularly monitoring iron levels and administering chelation Intravenously or orally places a treatment burden on patients. From an industry perspective, the lack of curative or disease-modifying treatment options limits growth potential. Pharmaceutical companies have fewer incentives to invest heavily in drug research and development for a rare disease with only supportive care options available. Overall, the unmet medical need of a cure drives both patient and industry interest in novel therapies that can target the underlying pathology.
Market Opportunity: Development of Disease-Modifying Therapies to Create Novel Opportunities.
There is promising opportunity in the developmesnt of disease-modifying therapies for alpha thalassemia that target the root cause of ineffective hemoglobin production. Two drugs, Mitapivat and Luspatercept, are in late-stage clinical trials investigating their potential to reduce transfusion dependency. Mitapivat activates pyruvate kinase which could restore red blood cell health and function. In clinical studies so far, it has demonstrated reductions in transfusion burden for certain patients. Luspatercept works by binding and inhibiting proteins responsible for ineffective erythropoiesis, thereby enabling normal red blood cell maturation. Its approval for beta-thalassemia has laid the foundation for studies in alpha thalassemia. If successful, these therapies may help improve clinical outcomes by enabling fewer transfusions and reducing iron overload management over the long term. Their approval could reinvigorate the market by fulfilling the critical need for curative or disease-modifying treatments for alpha thalassemia patients.
Prescribers preferences of Alpha Thalassemia Market
Alpha thalassemia is typically treated along two main lines of treatment - first-line and second-line. In first-line treatment, most prescribers prefer hydroxyurea for mild to moderate cases. Hydroxyurea, marketed under brand names Hydrea and Droxia, works to reduce abnormal hemoglobin proliferation and manage anemia symptoms.
For more severe cases requiring regular blood transfusions, prescribers commonly choose chelator therapy in addition to transfusions. Deferasirox (Exjade) and deferiprone (Ferriprox) are preferred iron chelator options prescribed to prevent hemochromatosis caused by excess iron absorption from regular transfusions.
If a patient does not respond adequately or becomes transfusion-independent, prescribers consider second-line treatments. Bone marrow transplant is recommended for eligible patients to definitively treat the disease. However, due to risks involved, prescribers thoroughly evaluate transplant suitability based on age, disease stage and availability of a matched donor.
For patients not eligible for transplant, prescribers prescribe Luspatercept-aamt (Reblozyl), a newly approved erythroid maturation agent, in combination with best supportive care including transfusions and iron chelation. Reblozyl can reduce transfusion dependency in some patients.
Overall prescribers seek treatments that alleviate anemia symptoms, reduce transfusion requirements, and consider individual patient factors like severity, progression rate and response to prior therapies. This helps determine the most suitable line and regimen of treatment for each alpha thalassemia case.
Treatment Option Analysis of Alpha Thalassemia Market
Alpha thalassemia has different levels of severity depending on the number of defective alpha globin genes. Treatment plans are tailored based on the specific type. In mild forms, no treatment may be needed as patients are often asymptomatic. In moderate forms, the first line of treatment is folic acid supplementation, which helps promote red blood cell production. For severe forms, regular blood transfusions are required to manage anemia symptoms. The preferred treatment is red blood cell transfusions every 2-5 weeks depending on the individual. This helps reduce other complications by maintaining consistent hemoglobin levels. Chelation therapy using deferoxamine is also given to remove excess iron from previous transfusions. For the most severe type called HbH disease, bone marrow transplant is the only curative option. However, it comes with risks and a perfect donor match is needed. The transplantation replaces defective bone marrow with healthy donor cells. Medications like busulfan and cyclophosphamide are provided as pre-transplant conditioning to eliminate defective cells.
For patients who cannot undergo transplant, the management focuses on lifelong blood transfusions along with chelation therapy to avoid serious organ damage from iron overload. Close monitoring helps determine optimal transfusion schedules tailored to each patient. Overall, a multidisciplinary care approach is needed to effectively manage the condition and improve quality of life based on the severity level.
Key winning strategies adopted by key players of Alpha Thalassemia Market
Adoption of Gene Therapy: Bluebird Bio adopted a gene therapy strategy for Alpha Thalassemia in 2019. They developed LentiGlobin, a gene therapy treatment targeting the underlying genetic defect that causes Alpha Thalassemia. In clinical trials, LentiGlobin demonstrated significant transfusion independence in patients, eliminating or reducing their dependency on regular blood transfusions. This innovative gene therapy approach transformed the treatment landscape for Alpha Thalassemia. It received marketing approval in Europe in 2019, making it the first approved gene therapy for the disease.
Acquisition Strategy: Celgene pursued an acquisition strategy, acquiring Agios Pharmaceuticals in 2018 for USD7 bn. Agios had an experimental drug called mitapivat in phase 3 trials for Thalassemia. This allowed Celgene to gain access to a promising late-stage pipeline asset for Alpha Thalassemia. In 2020, mitapivat was approved by the FDA for Thalassemia, becoming the first approved disease modifying therapy to reduce transfusion burden for non-beta thalassemia patient populations.
Collaborations and Partnerships: Global Blood Therapeutics partnered with specialty pharmacies and non-profits focused on rare diseases to expand access to their FDA approved product Oxbryta for SCD. They provided financial aid and co-pay assistance programs for eligible Alpha Thalassemia patients. This helped overcome affordability barriers and drive uptake of Oxbryta, which reduces anemia and dependence on blood transfusions. Prescriptions for Oxbryta in the Alpha Thalassemia indication doubled in 2021 compared to 2020.
These examples demonstrate successful strategies such as gene therapy innovation, targeted acquisitions, and partnerships to expand access and reimbursement.
Segmental Analysis of Alpha Thalassemia Market
Insights, By Type of Disease: The Growig Need for Transfusion-Dependent Alpha Thalassemia (TDT) is Evident.
By Type of Disease, Transfusion-Dependent Alpha Thalassemia (TDT) is expected to contribute 52.5% in 2024 owing to the critical nature of care required by patients. TDT is a severe form that requires lifelong regular blood transfusions for survival. Patients experience severe anemia and other complications if transfusions are not properly managed. This makes treatment non-negotiable and a primary healthcare priority. Families of TDT patients face immense medical and financial burdens to sustain long-term transfusion therapies. Psychosocial challenges are also prevalent due to the dependence on medical services. As a result, healthcare systems are prompted to optimize access to quality-matched blood and invest in transfusion programs to prevent mortality.

Insights, By Treatment Type, Iron Chelation Therapy is Poised to Register a Remarkable Growth in the Coming Years.
By Treatment Type, Iron Chelation Therapy is expected to contribute 36.1% in 2024 owing to its widespread adoption as a frontline intervention. Transfusions effectively address anemia but introduce iron overload as an inevitable side effect if left unmanaged. Chelation therapy aims to bind and remove excess iron accumulated in vital organs through multiple daily dose schedules. Desferrioxamine via subcutaneous infusion remains a standard treatment globally despite its inconvenience. Newer oral agents like deferiprone and deferasirox have gained preference due to better compliance from oral administration. As life-expectancies of TDT patients rise with quality care, long-term iron control holds importance to avert complications and maximize wellbeing.
Insights, By End-user, Pediatric is Expected to Witness a Remarkable Growth in the Forecast Period.
By End-user, Pediatric contributes the highest share of the market owing to TDT often being diagnosed at birth or in early childhood. Symptoms manifest from infancy, necessitating rapid intervention. Healthcare utilization is intensive from screening and diagnostic evaluations to longitudinal transfusion therapies throughout development. Aside from medical requirements, pediatric patients also need extensive education, counseling and coordinated multidisciplinary support to ensure adherence. Timely management amid growth stages aims to optimize physical, cognitive and social milestones. While adult care remains essential, pediatric demand takes precedence to address the disease from its onset and minimize long-term consequences.
Additional Insights of Alpha Thalassemia Market
Alpha Thalassemia is a genetic disorder that affects the production of hemoglobin, leading to severe anemia and other complications. While the disease is more prevalent in developing countries, the US and Europe are seeing increasing cases due to population diversity. The market for Alpha Thalassemia is largely driven by traditional treatments such as blood transfusions and iron chelation therapies, but the emergence of new therapies like Mitapivat and Etavopivat offers hope for non-transfusion-dependent patients. These new therapies aim to address the underlying genetic causes and improve red blood cell health, which could significantly reduce the need for frequent transfusions and improve quality of life. Despite the high potential of these treatments, challenges remain, including high costs, lack of awareness, and limited access to advanced therapies in developing regions. As research progresses and more therapies reach the market, the Alpha Thalassemia landscape is expected to evolve, with better outcomes for patients and significant growth in the market.
Competitive overview of Alpha Thalassemia Market
The major players operating in the Alpha Thalassemia Market include Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc, Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc, Astrazeneca, F.Hoffman La Roche and Jansen Pharmaceutical.
Alpha Thalassemia Market Leaders
- Agios Pharmaceuticals
- Novo Nordisk
- Bristol-Myers Squibb
- Forma Therapeutics
- Silence Therapeutics
Alpha Thalassemia Market - Competitive Rivalry, 2024

Alpha Thalassemia Market
(Dominated by major players)
(Highly competitive with lots of players.)
Recent Developments in Alpha Thalassemia Market
- In January 2024, Agios Pharmaceuticals announced positive Phase III trial results for Mitapivat in non-transfusion-dependent Alpha Thalassemia patients, showing a significant increase in hemoglobin levels. Regulatory approval is expected in 2025.
- In September 2022, Novo Nordisk acquired Forma Therapeutics and is now developing Etavopivat, an investigational oral drug that aims to reduce the need for transfusions by improving red blood cell health in Alpha Thalassemia patients.
Alpha Thalassemia Market Segmentation
- By Type of Disease
- Transfusion-Dependent Alpha Thalassemia (TDT)
- Non-Transfusion Dependent Alpha Thalassemia (NTDT)
- By Treatment Type
- Iron Chelation Therapy
- Blood Transfusions
- Bone Marrow Transplants
- By End-user
- Pediatric
- Adult

Would you like to explore the option of buying individual sections of this report?
Frequently Asked Questions :
How Big is the Alpha Thalassemia Market?
The Global Alpha Thalassemia market is estimated to be valued at USD 4.4 bn in 2024 and is expected to reach USD 9.1 bn by 2031.
What will be the CAGR of the Alpha Thalassemia Market?
The CAGR of the Alpha Thalassemia Market is projected to be 9.10% from 2024-2031.
What are the key factors hampering the growth of the Alpha Thalassemia Market?
The lack of approved therapies for alpha thalassemia, with patients relying on traditional treatments like blood transfusions and iron chelation and high costs and limited access to advanced treatments, particularly in developing nations where the prevalence is higher are the major factors hampering the growth of the Alpha Thalassemia Market.
What are the major factors driving the Alpha Thalassemia Market growth?
The increasing awareness and research in genetic disorders such as alpha thalassemia, leading to earlier diagnosis and improved treatment approaches and promising pipeline therapies like Mitapivat, Etavopivat, and Luspatercept are expected to drive the market, particularly for non-transfusion-dependent cases are the major factors driving the Alpha Thalassemia Market.
Which is the leading Type of Disease in the Alpha Thalassemia Market?
Non-Transfusion Dependent Alpha Thalassemia (NTDT) is the leading Type of Disease segment.
Which are the major players operating in the Alpha Thalassemia Market?
Agios Pharmaceuticals, Novo Nordisk, Bristol-Myers Squibb, Forma Therapeutics, Silence Therapeutics, Calimmune Inc., CRISPR Therapeutics, Editas Medicine Inc, Errant Gene Therapeutics LLC, Gamida Cell Ltd, Gilead Sciences Inc, Astrazeneca, F.Hoffman La Roche, Jansen Pharmaceutical are the major players.